Protein Preparation
Introduction
A complete and reasonable protein structure file is the starting point for protein property analysis and other CADD calculations, such as active site prediction and docking.
The Protein Preparation module of the Hermite platform offers functionalities for protein structure processing, including structure selection, completion of missing residues and side chains, determination of protonation states for polar residues, and optimization of the hydrogen bond network.
This tutorial will use 1iep[1] as an example to demonstrate protein preparation using the Protein Preparation module on the Hermite platform.
1. Protein Preparation
1.1 Accessing Protein Preparation
Navigate to Protein Preparation through the general menu bar on the left: Function → General → Protein Preparation.
1.2 Select Structure
To acquire the structure of 1iep using its PDB ID: Click the "PDB" button, which opens the "Get PDB" window. In this window, enter "1iep" in the "PDB ID" field and click "Import." The structure of 1iep will then be imported into the protein preparation task and displayed in the 3D Workspace interface.
Note:For operating instructions on more protein selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
1.3 Select Polymer/Water(s)/Other Groups to Keep
1iep is a homodimer structure. This tutorial only prepares protein for Chain A. The specific steps are as follows:
lick "Next" in the Protein Preparation panel on the right hand side;
In "Polymer to Keep", select Chain A;
In "Select Water(s) to Keep" select "Keep Water(s) around Hetatms" and check "A: STI 201" and set it to 6 Å;
In "Select Other Groups to Keep": Do not select any other groups in this case.
Click "Next"
Explannation of the different options are shown in the table below:
| Parameter | Description | |
| Alternative Position | If there are atoms with multiple coordinates in the protein, the desired position can be selected based on the probability of the position appearing | |
| Select Polymer to Keep | Select the chain that needs to be preserved | |
| Select Water(s) to Keep | Keep Water(s) around Hetatms | Select to retain water molecules around non-protein molecules, with customizable range |
| Keep All Water(s) | Retain all water molecules | |
| Delete All Water(s) | Remove all water molecules | |
| Select Other Groups to Keep | Select other groups that need to be retained |
1.4 Repair Missing Residues and Parameter Settings
In "Select Missing Residues to Repair", select all missing residues of Chain A and keep all other settings as default.
Click "Submit.
| Parameter | Note |
| Select Missing Residues to Repair | Repair can be performed based on the sequence-related data in the .pdb file. You can choose to upload a .fasta file to fix the missing information using the "Fill Missing Residue" option. |
| Add Missing Side Chains | |
| Add Hydrogens | It is recommended to check this option. |
| Modify Protonation State | Adjust the pH of the environment to achieve the protonated state of the protein |
| Optimize Hydrogen Bonding Network | It is recommended to check |
| Energy Minimization | Adjust according to the specific system |
2. Viewing Protein Preparation Results
2.1 Accessing Task Results
To access the results (usually becomes available within 1 min after submission of this task) , use the left general menu bar and select Job → Locate "1iep_prep" → Click "Show" (first icon under Operation) to display the task results.
The task can be found either by searching for the Job Name or by filtering through Job Type.
2.2 Displaying and Comparing Protein
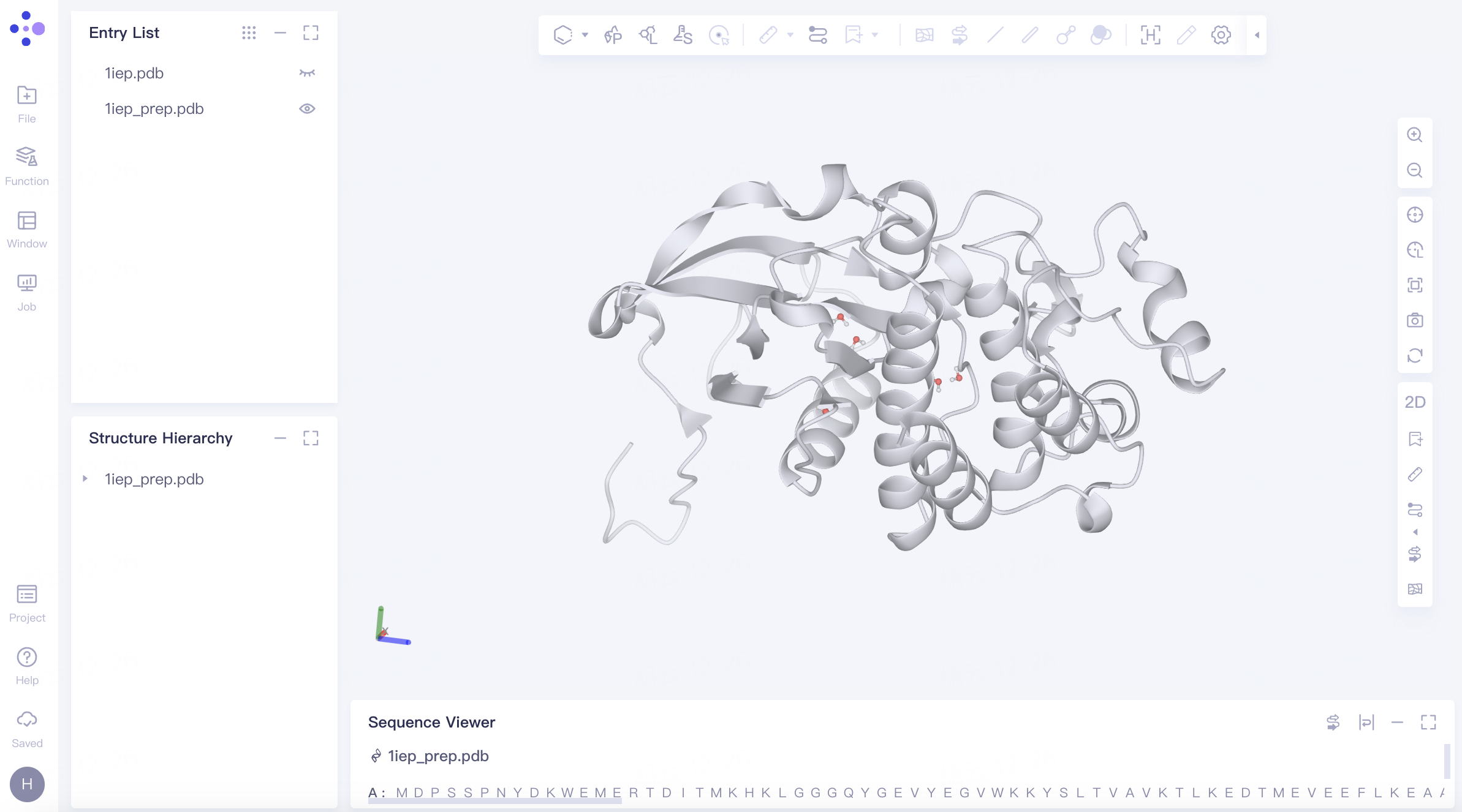
The processed protein "1iep_prep" is displayed in the 3D Workspace.
Protein Coloring: In the "Entry List," double-click "Show" (the eye icon next to "1iep.pdb") to fix the display of the protein → Expand "1iep.pdb" under "Structure Hierarchy", right-click on "Protein", select "Style", choose a color for "Ribbon" in the pop-up Style window, and click "Apply".
Comparing with the Original Crystal Structure: In the "Entry List," click "Show" (the eye icon next to "1iep_prep.pdb") next to "1iep_prep.pdb" to fix the display of the prepared protein structure. Rotate the protein in the 3D Workspace to compare the conformation before and after protein preparation. It is observed that the missing residues are filled and there is no significant difference in conformation before and after protein preparation.
3. References
[1] Nagar B, Bornmann WG, Pellicena P, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002;62(15):4236-4243.