Docking
Introduction
Molecular docking is a key technique in developing small molecule drugs. It predicts the binding pose, affinity, and active sites between drug molecules and proteins, providing a theoretical basis for designing, screening, and optimizing pharmaceuticals.
Our team developed Uni-Dock, a GPU-accelerated molecular docking program, to efficiently screen large databases containing tens to hundreds of millions of molecules. Uni-Dock outperforms the single CPU core operation of AutoDock Vina, boasting over 1000 times faster speed and greater accuracy than existing GPU-accelerated docking programs like AutoDock-GPU and Vina-GPU[1]. We have further refined the docking algorithm and improved the user interface, adding four Constrained Docking methods: H-Bond, Maximum Common Structure, Substructure, and Shape. Leveraging the Uni-Mol pretrained model, Uni-Dock delivers enhanced docking pose accuracy.
We will use the following case study to demonstrate Uni-Dock.
The human epidermal growth factor receptor (EGFR) is linked to small cell lung cancer, breast cancer, and glioblastoma. EGFR tyrosine kinase inhibitors are now standard treatments for non-small cell lung cancer (NSCLC), breast cancer, and cases with EGFR mutations or resistance. 8A2D is a co-cystal structure of EGFR mutant and compound EGFRai 57, featuring a resolution of 1.11 Å. EGFRai 57 demonstrated potential in treating NSCLC, either used alone or combined with osimertinib[2]. This tutorial uses Hermite's Docking module to perform Redocking on the 8A2D structure
The required documents for this tutorial are as follows:
1. Protein and Ligand Check
1.1 Importing Protein and Ligand Files
Navigate to "File" → "Import Structure".
In the "Import Structure" window, click "Select Files", choose the local files "8A2D_Lig_prep" and "8A2D_pro_prep", click "Import" to load the files into the 3D Workspace.
1.2 Displaying Protein-Ligand Interaction
In the Entry List, double-click the eye icon or right-click and select "Fix" to stabilize the display of proteins and ligands.
Click "Hydrogen Display Setting" in the 3D Workspace's top toolbar, select "Hide All Polar Hydrogen".
Use the "Interaction Toggle" in the 3D Workspace's top toolbar to check for interactions. If there's no interaction between the ligand and nearby water molecules, these water molecules can be excluded from further steps.
2. Docking
2.1 Accessing the Docking Function
Go to "Function" → "Virtual Screening" → "Docking".
2.2 Selecting a Prepared Protein
Click "Select from 3D Workspace". In the pop-up "Select Structure" window, expand "8A2D_pro_prep.pdb" in the Structure Hierarchy, click "Protein", and then click "OK". The force field of the protein will be uploaded for validation. Once the status changes from "Processing" to "Valid" in the Valid column, click "Next".
Note: If the force field check fails, use the "Protein Preparation" button in the upper right corner to prepare the protein. For operating instructions on more protein selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
2.3 Selecting Prepared Ligands
Click "Select from 3D". In the pop-up "Select Structure" window, choose "8A2D_Lig_prep" from the Structure Hierarchy → the selected ligand will appear in the "Select Ligands" window, click "OK".
Click "Next".
Note: Ensure that ligands for the Docking task are prepared, including hydrogenation, protonation, and energy minimization. For operating instructions on more ligand selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
2.4 Pocket Setting
Choose the first icon "Select Ligand in the structure as center" and choose "8A2D_Lig_prep_A MOL 1" to create the pocket based on the ligand. Keep default parameters and click "Next".
Note:For operating instructions on more pocket setting methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
2.5 Constrained Docking
There is no need to do constrained docking here, so click "No, skip this step" to skip this step.
Note: refer to the table below for constrained docking operations:
| Types of Constrained Docking | Operation | Interface display | Note |
| H-Bond | 1) Select "H-Bond" and choose the reference ligand displayed in "Ligand Showed in 3D Workspace". Click "Confirm". 2) In the 3D Workspace window, select atoms in the acceptor that interact via hydrogen bonds with the ligand. These atoms will be automatically categorized into the hydrogen bond acceptor and donor list on the right. | 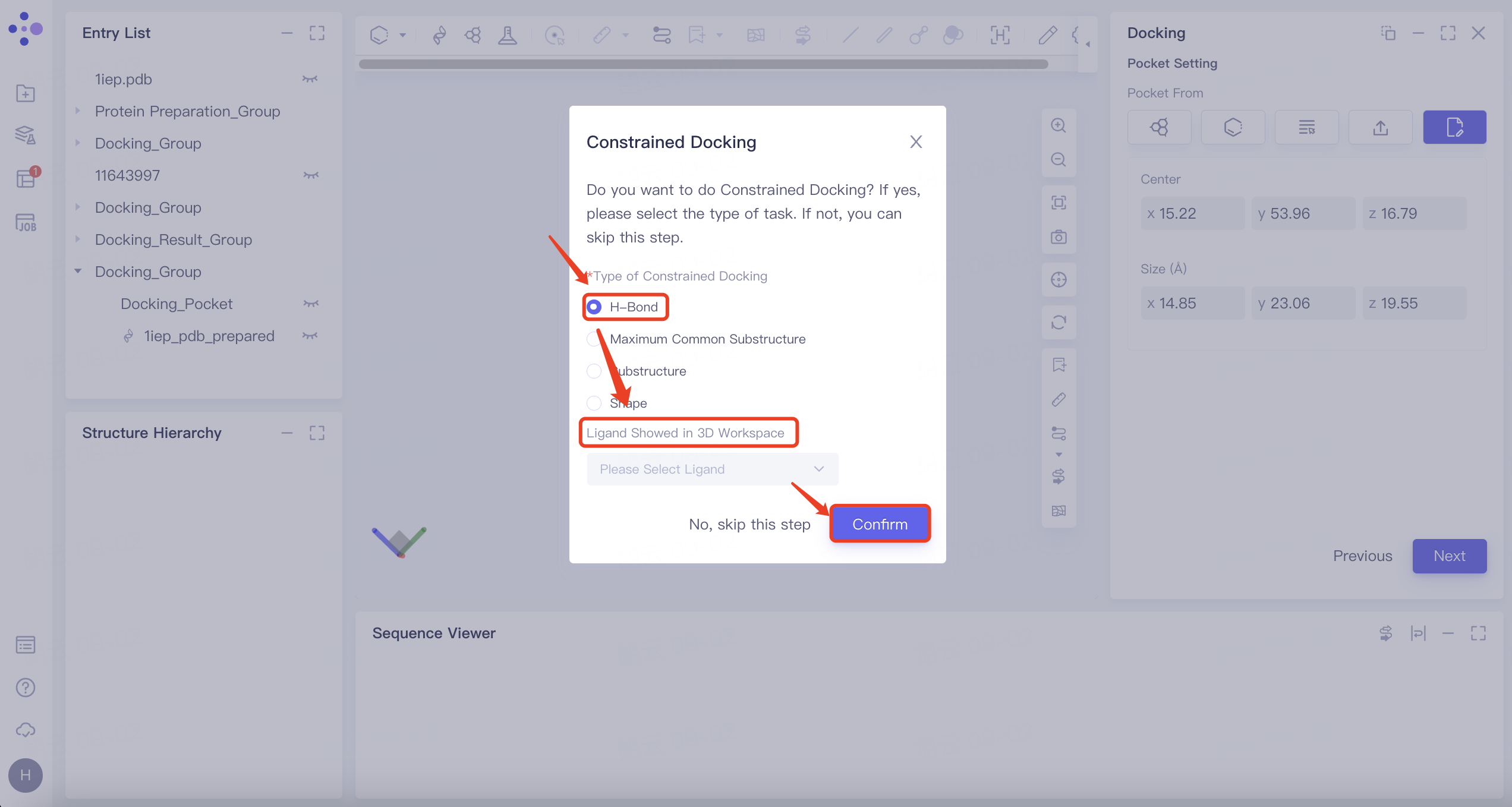 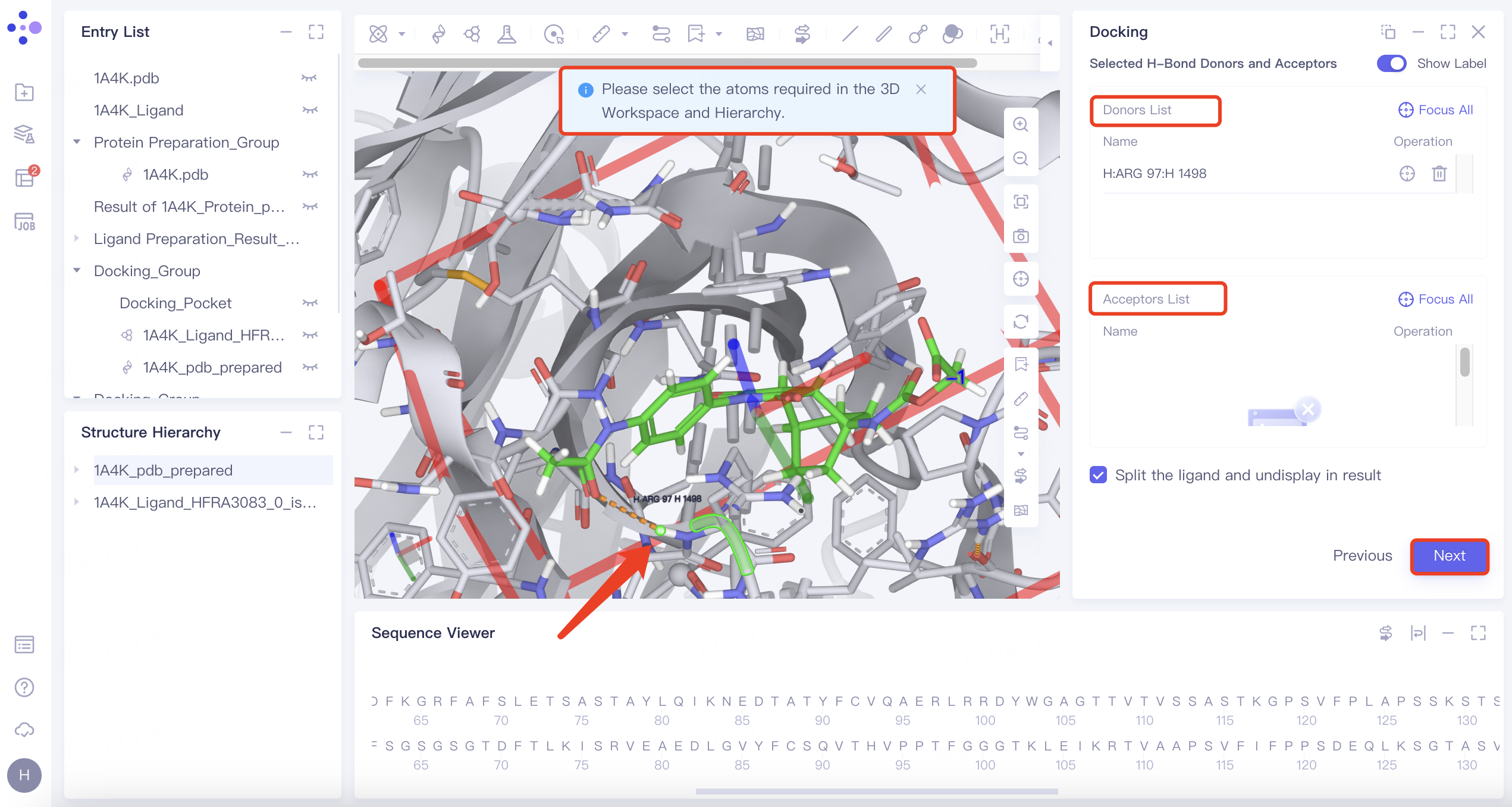 | 1) When certain hydrogen bonds formed between ligands and acceptors are associated with molecular activity, new molecules with potential activity can be screened by H-bond constrained docking. 2) The orange dashed line indicates the hydrogen bond interaction that currently exists between the ligand and the acceptor. |
| Maximum Common Substructure | 1) Click the "Maximum Common Substructure" button and then click "Confirm". 2) Select the reference ligand (the ligand with the desired binding pattern to the receptor). The ligand will undergo force field inspection, and upon passing, the "Valid" status will be displayed in the Valid column. Simultaneously, the 2D molecular diagram of the ligand will appear in the "Structure View" window. | 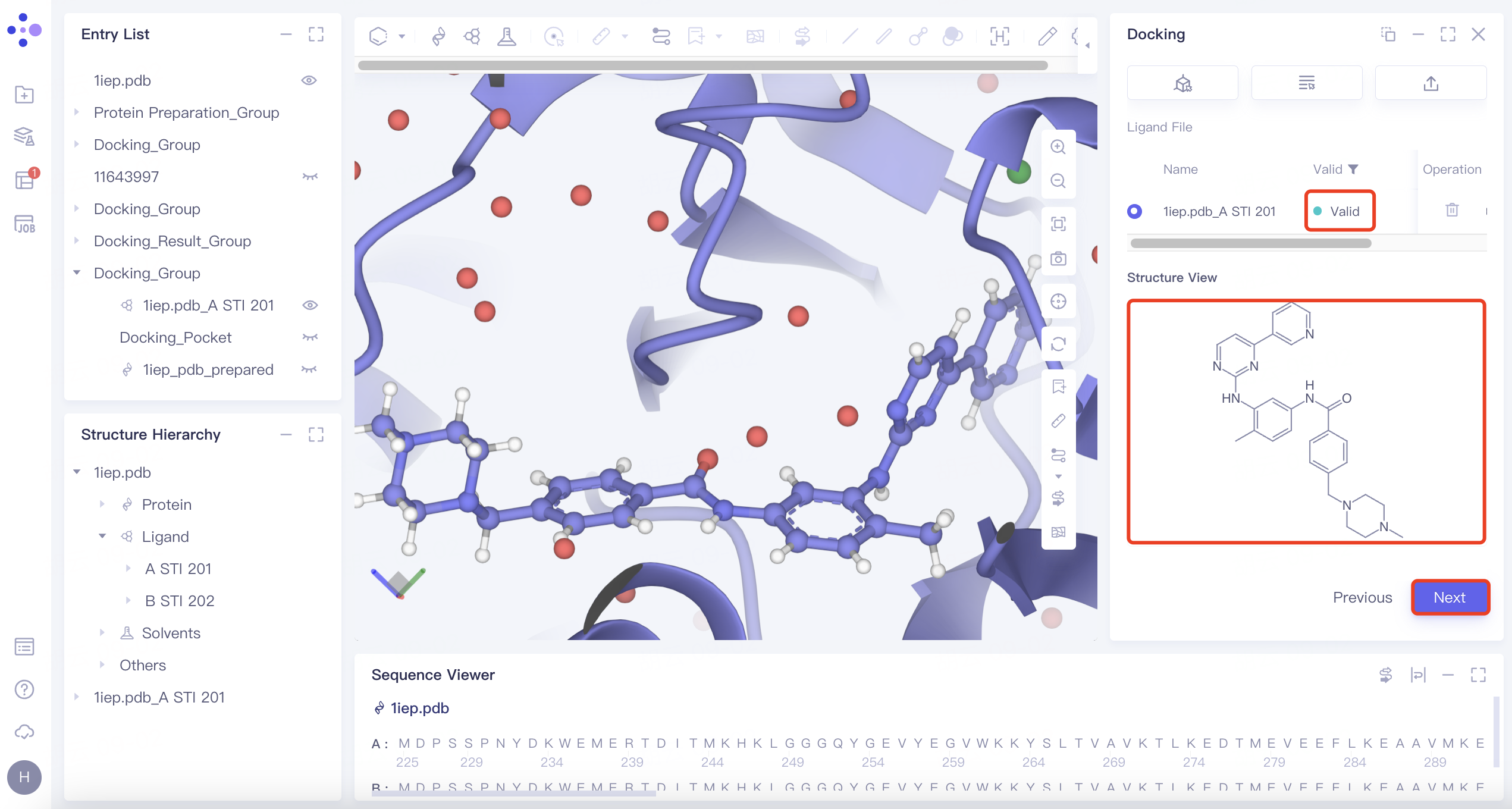 | Use "Maximum Common Substructure" when you have identified a ligand with an effective binding pose to a receptor and wish to identify other molecules that share this binding pose. |
| Substructure | 1) Click the "Substructure" button, then click "Confirm". 2) Select the reference ligand (a ligand with specific substructures). Upload the ligand for force field inspection. Once it passes the inspection, "Valid" will be displayed in the Valid column. The 2D molecular diagram of the ligand will appear in the Structure View window. After selecting the substructure, click "Save" to store the substructure information. | 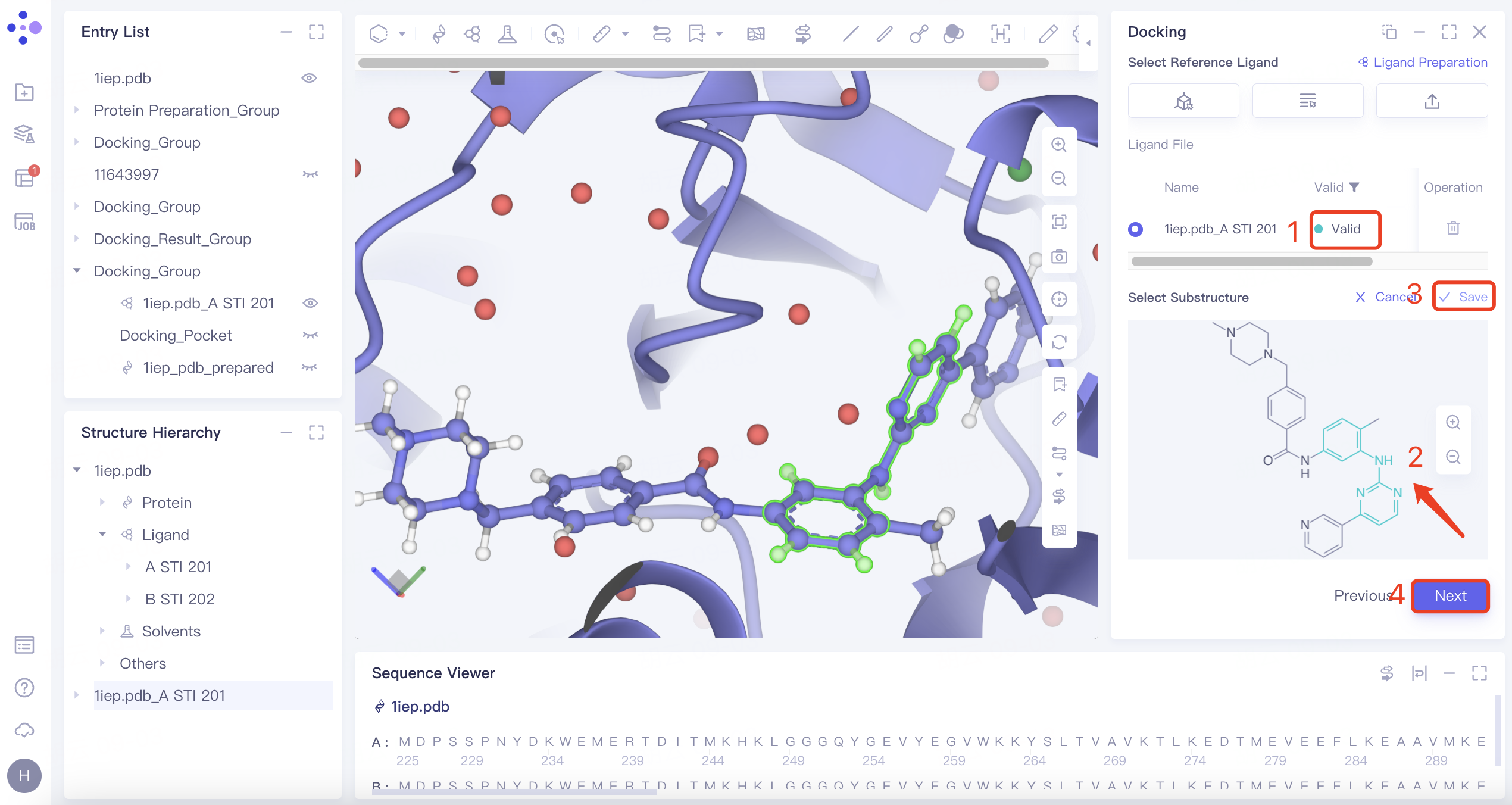 | The Substructure option facilitates molecular docking by focusing on substructure screening. This approach is used to search for ligands that contain specific substructures, aiding in targeted docking processes. |
| Shape | 1) Click the "Shape" button, then click "Confirm". 2) Select the reference ligand (a ligand with a specific shape). Upload the ligand for force field inspection. Once it passes the inspection, "Valid" will be displayed in the Valid column. Define the shape by expanding the van der Waals radius of the reference ligand atoms. Click "Sync to 3D" on the right to visualize the shape in the 3D Workspace window. | 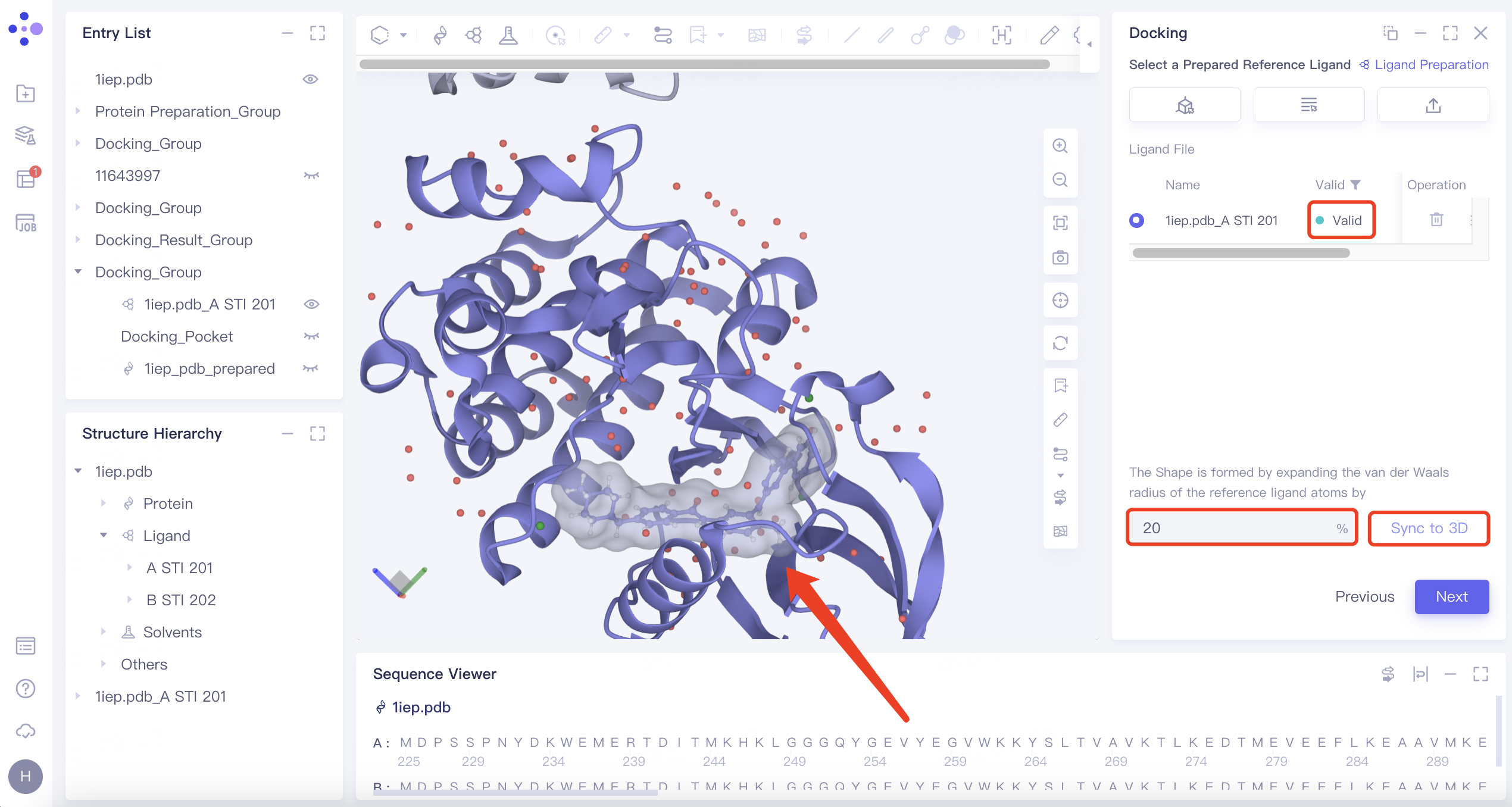 | 1) Shape-based virtual screening is crucial for skeleton transition, biological isomer substitution, and virtual library design. The "Shape" option enables shape-based virtual screening. 2) or example, setting the ratio to 20% means expanding the van der Waals radius of the original molecule by 20% to create a new volume. |
2.6 Confirmation and Setting
Verify the selected protein, ligands, and docking box.
Scoring Function: Set to "Vina".
Search Mode: Choose "Detailed".
Number of Results to Keep: Select "All".
Multi Binding Poses: Enable "Keep Multi Binding Poses for Each Ligand". Set "Number of Binding Pose" to 20 and "Energy Range" to 9.
Pose Optimization: Activate "Use Uni-Mol to optimize the resulting ligands' pose(s)".
Rescoring Function: Select Gnina for rescoring.
Task Submission: Review the calculation cost under "Calculation". Name the task "8A2D_Gnina" in the "Job Name" field. Click "Submit" to initiate the task.
| Settings | Description | Note |
| Scoring Function | Currently supports Vina, Vinardo, AutoDock4. | Vina is most commonly used. |
| Search Mode | Fast:exhaustiveness 128 & max_step 20. | Different molecular docking computational complexities are set for each search mode, with 'Detail' offering the most comprehensive coverage of the search space and the highest accuracy, followed by 'Balance'. |
| Balanced:exhaustiveness 384 & max_step 40. | ||
| Detailed:exhaustiveness 512 & max_step 40. | ||
| Keep Multi Binding Poses for Each Ligand (multiple conformations per ligand) | 1) Number of Binding Pose: supports up to 100 conformations. 2) Energy Range (kcal/mol): This represents the maximum energy difference allowed from the optimal combination model, with a supported range of 1 to 9 kcal/mol. | —— |
| Use Uni-Mol for pose optimization | Optimize the pose when docking. | A constrained docking based on Machine Learning methods. |
| Rescoring | Re-score the docking conformation. Re-score function include Vina, Vinardo, GNINA, KarmaDock Affinity, RTMScore, Uni-Mol Affinity, Uni-Score. | Gnina is recommended. |
3. Result analysis
3.1 Job Entry
Navigate to "Job" and search for "8A2D_Gnina_Rescore" in the Job List interface. Click "Show".
Note: The task "8A2D_Gnina_Rescore" is generated because "Rescoring" was selected.
3.2 Result List
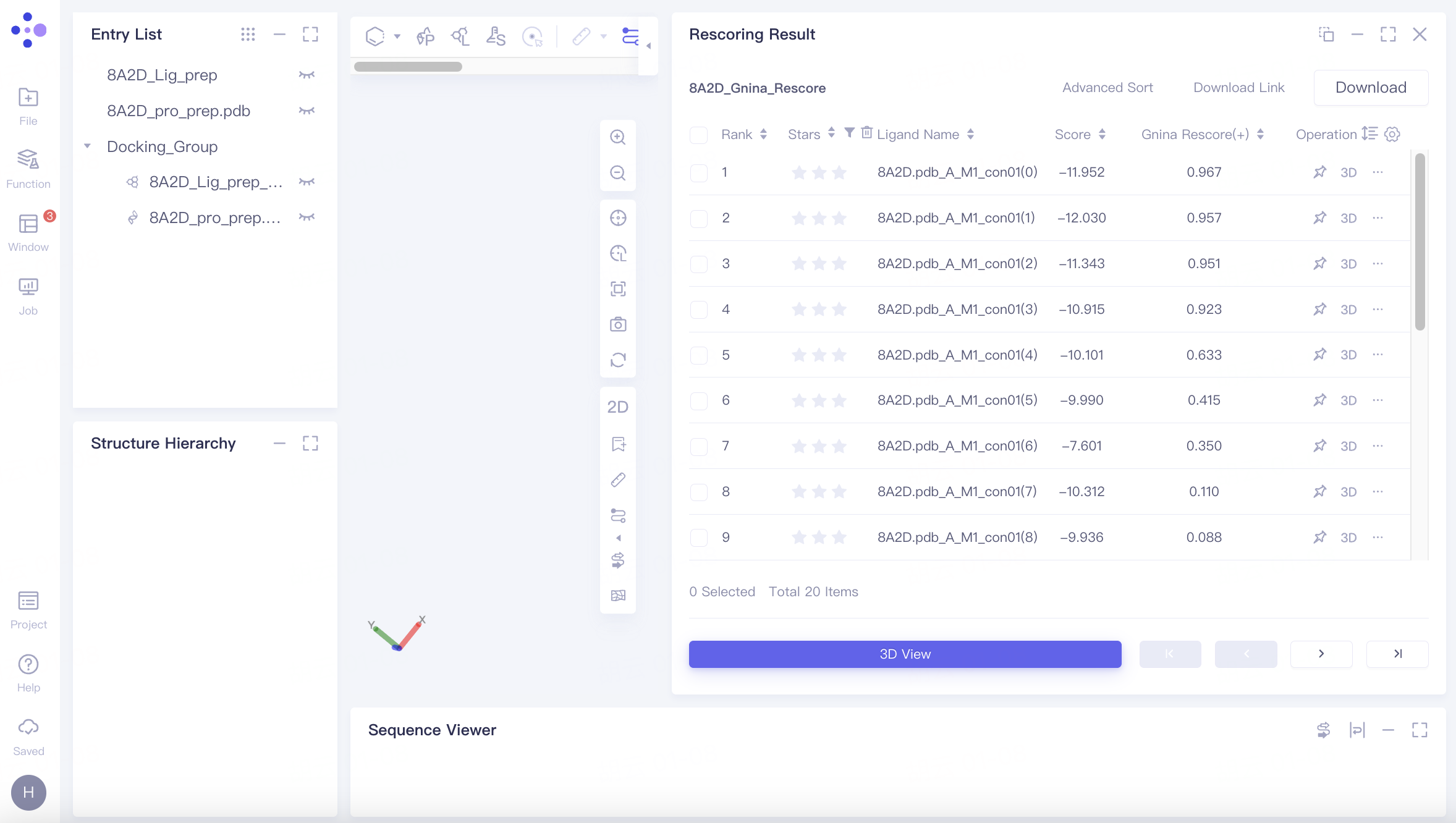
Docking results are sorted by rescore by default. The highest Gnina score observed is 0.967.
Note: A "(+)" symbol indicates higher scores are better, while a "(-)" symbol indicates lower scores are better.
3.3 Conformation Reproduction
In "Entry List", double-click the eye icon next to "8A2D_Lig_prep" to fix the display of the reference ligand.
Select the ligand in the 3D Workspace, right-click, choose "Style", select a color under "Atom", and click "Apply". The reference ligand in the 3D Workspace will be highlighted in the selected color.
For the top-scoring ligand, click "3D" in the Operation column to display its docking results with the receptor in the 3D Workspace window.
Observe the ligand pose before and after docking in the 3D Workspace window. The Redocking process accurately reproduces the original binding pose, indicating the tool's effectiveness for virtual screening.
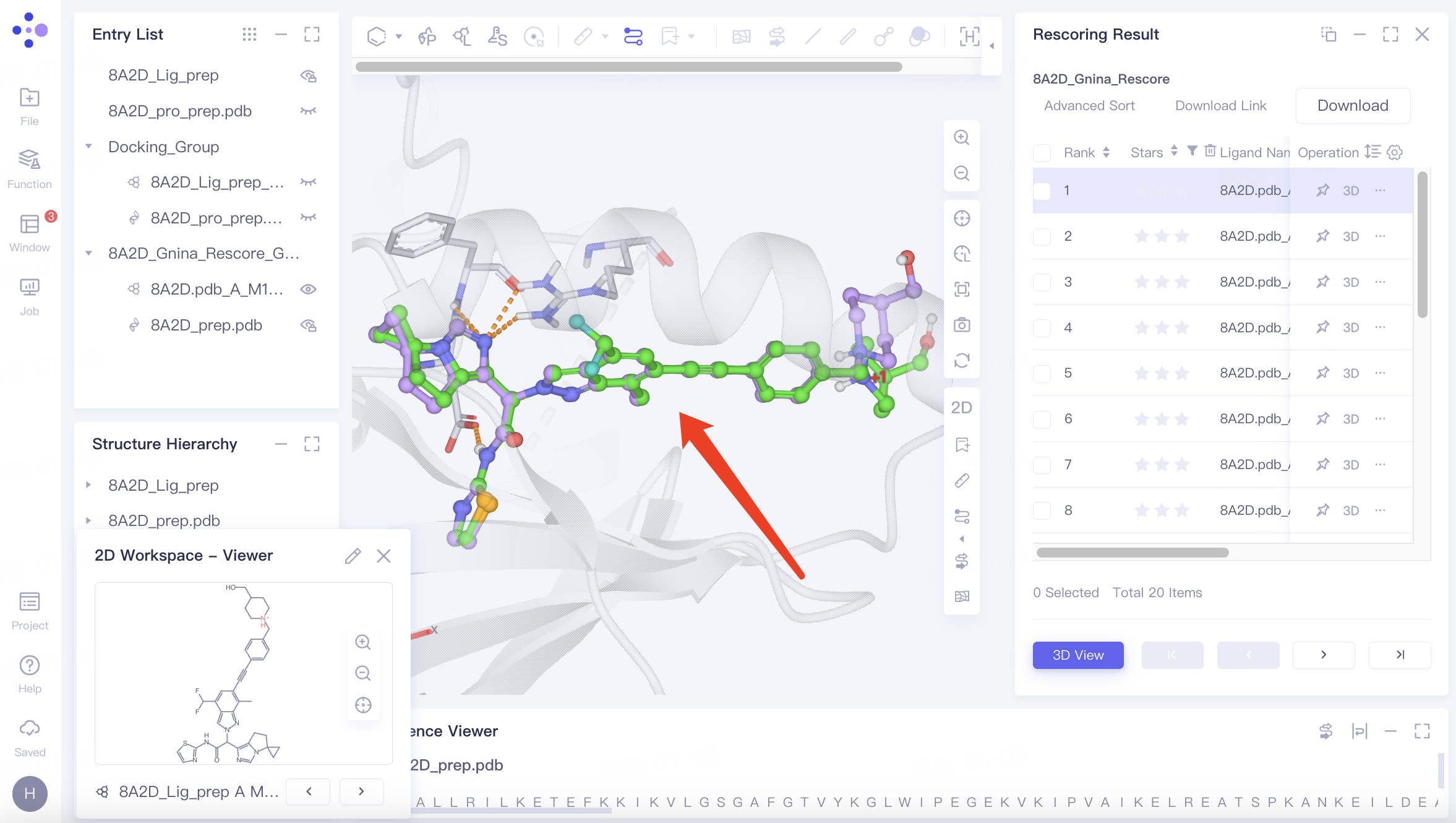
Use "↓" and "↑" keys to cycle through different docking results.
3.4 Screening for Specific Interactions
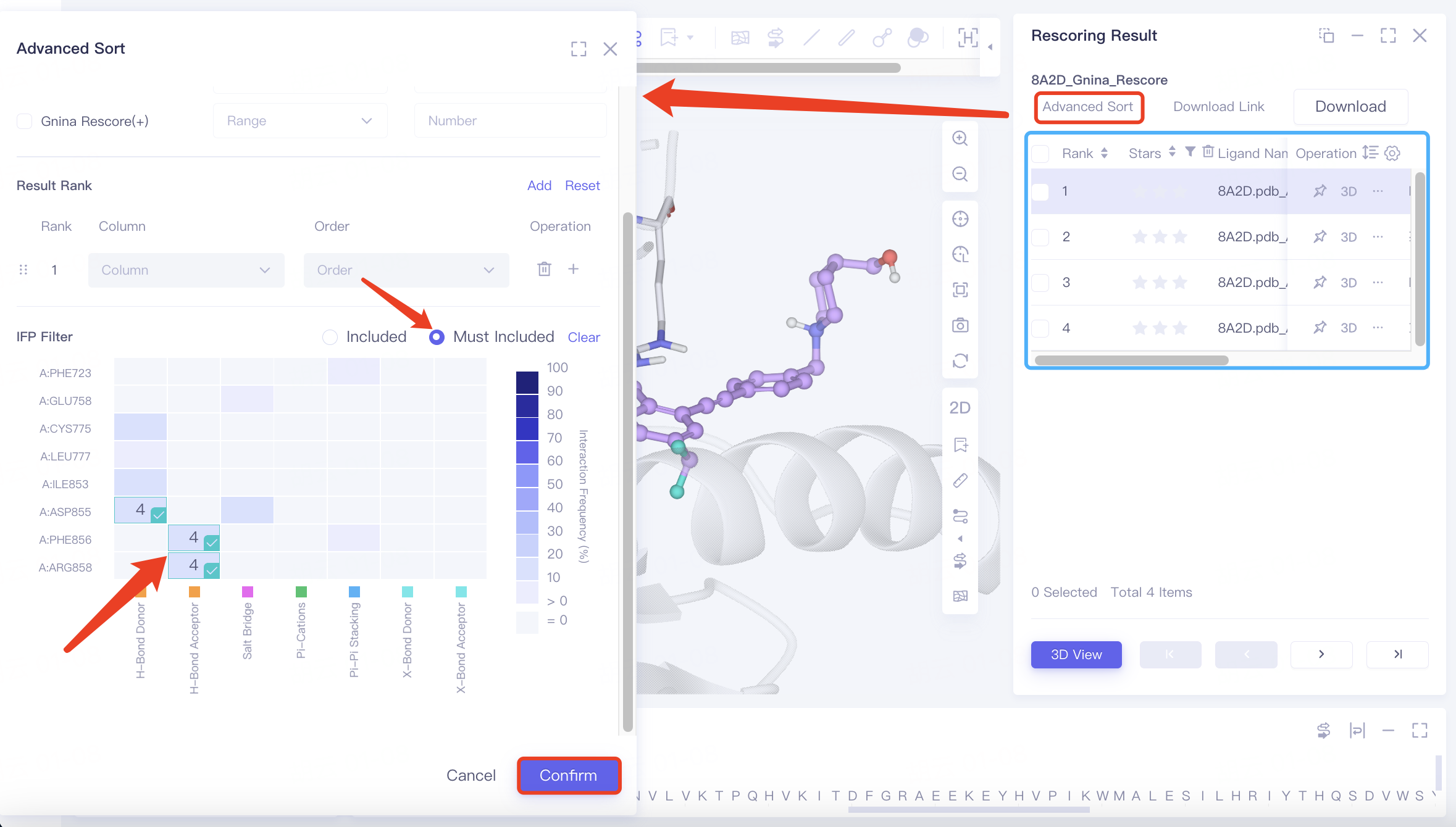
In the original conformation, hydrogen bonds are observed between the ligand and protein residues ARG 858 A, PHE 856 A, and ASP 855 A. To refine the search, use "Advanced Sort" to filter results.
Click "Advance Sort", find "IFP Filter" in the interface, select the three amino acid residues on the heatmap, choose "Must Include", and click "Confirm".
After applying the filter, four specific docking results are identified.
- Advanced Sorting Options:
- Result Filter: Refine docking results by specifying ranges for Star, Score, and Rescore values.
- Result Rank: Organize docking results by Ligand Name, Star, Score, or Rescore.
- IFP Filter: Filter docking results based on ligand-protein interactions:
- Include/Must Include:
"Include" retains results with any selected interactions.
"Must Include" retains results only if all selected interactions are present. - Heat Map:
The x-axis represents interaction types, while the y-axis shows residue names and IDs. The color intensity indicates the frequency of ligand-residue interactions in the docking results.
Hovering over a heatmap cell displays the number of results containing that specific interaction.
- Include/Must Include:
- Result Filter: Refine docking results by specifying ranges for Star, Score, and Rescore values.
4. References
[1] Yu Y, Cai C, Wang J, et al. Uni-dock: Gpu-accelerated docking enables ultralarge virtual screening[J]. Journal of Chemical Theory and Computation, 2023, 19(11): 3336-3345.
[2] Obst-Sander U, Ricci A, Kuhn B, et al. Discovery of novel allosteric EGFR L858R inhibitors for the treatment of non-small-cell lung cancer as a single agent or in combination with osimertinib[J]. Journal of medicinal chemistry, 2022, 65(19): 13052-13073.