Uni-MM PB/GBSA Structure
Introduction
Calculating the binding free energy between between ligands and protein receptors is a key step in drug discovery. The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) methods are widely used for this purpose. These techniques offer greater accuracy than most molecular docking scoring functions and are more efficient than free energy perturbation methods.
Our team has developed Uni-GBSA, an automated calculation suite for binding energy calculations, based on molecular dynamics simulations. Uni-GBSA streamlines the entire MM/GB(PB)SA calculation process, including topology preparation, structure optimization, and binding free energy calculations. It is designed to handle screening and ranking of millions of compounds in molecular libraries. Additionally, the team has rigorously tested parameters on the PDBBind-2011 refine-set, which includes 1864 protein-ligand complexes, to establish a robust set of MM/GB(PB)SA calculation parameters. This ensures reliable performance of Uni-GBSA's default settings across a wide range of systems. In the case study below, Uni-GBSA produced a satisfactory correlation with experimental binding affinity and outperformed AutoDock Vina [1].
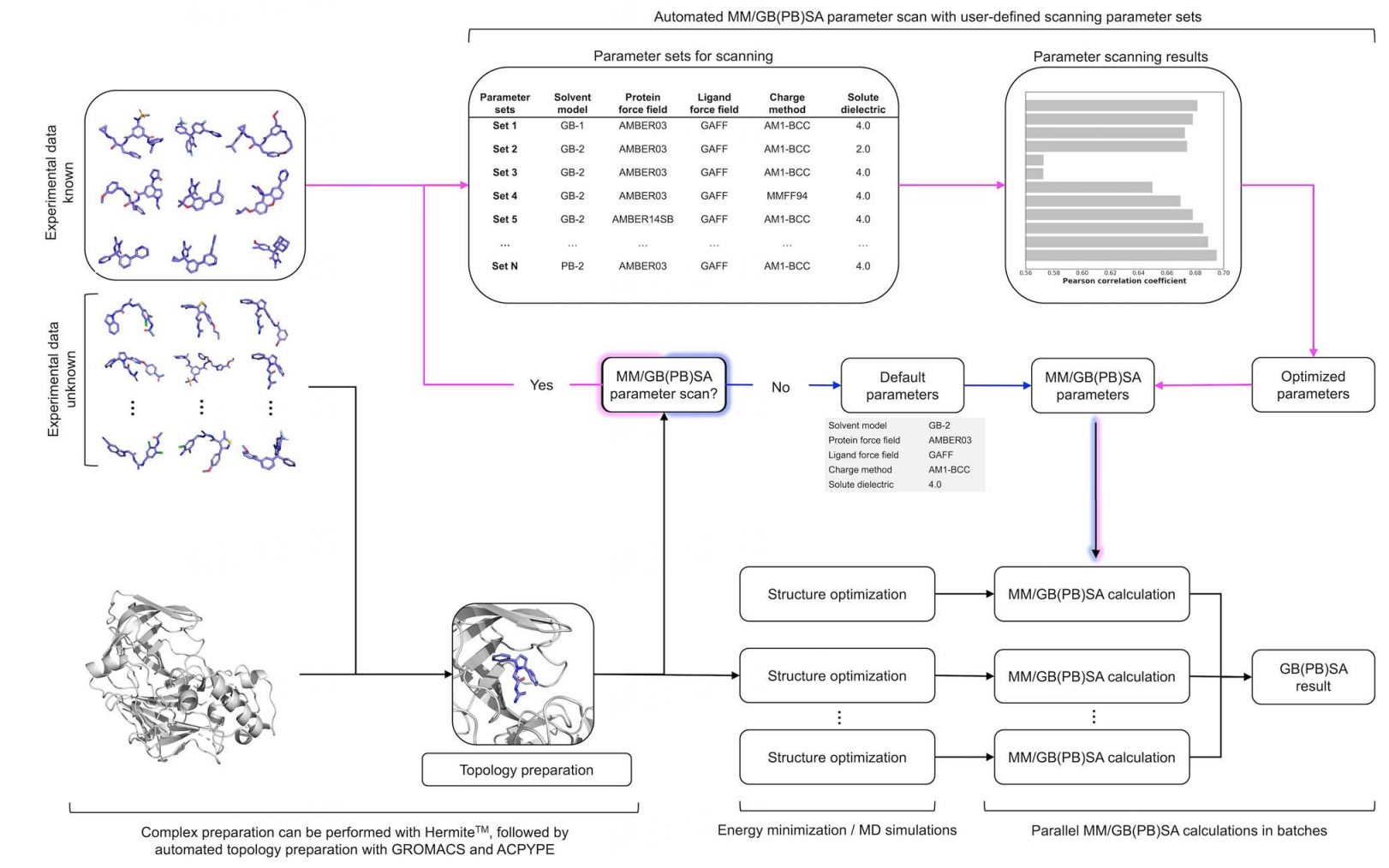
The Uni-GBSA tool is available on the Hermite platform, featuring an intuitive graphical interface for easy task submission and result analysis, and it supports large-scale computations for batch processing of millions of molecules.
In this tutorial, you will learn to use the Uni-MM PB/GBSA module on the Hermite platform to predict the binding free energy between the SARS-CoV-2 main protease and five computationally screened compound inhibitors [2]. This will help you understand the binding affinities of these inhibitors to the SARS-CoV-2 main protease, providing valuable insights into their potential as therapeutic agents.
Protein structure files used in this tutorial:
Compound structure files used in this tutorial:
The IC50 experimental values of the inhibitors used in this tutorial are:
| PDB ID | Compound | IC50 (μM) |
| 7O46 | 17 | 0.15 |
| 7NEO | 15 | 6.60 |
| 7B2J | 5 | 7.20 |
| 7B5Z | 6 | 38.50 |
| 7B77 | 8 | 79.30 |
1. Protein Preparation
1.1 Accessing Protein Preparation
Navigate to Protein Preparation through the general menu bar on the left: Function → General → Protein Preparation.
Note: For detailed protein preparation instructions, refer to "Protein Preparation."
1.2 Select Structure
Choose "Select File" → Upload the local file "7B2U.pdb."
7B2U.pdb" will be loaded into the Protein Preparation parameter setting panel.
For operating instructions on more protein selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
1.3 Select Polymer, Water(s), Other Groups to Keep
Under "Select Polymer to Keep", choose Chain B. Under "Select Water(s) to Keep", select "Delete All Water(s)" and click Next (there are no other groups in this protein structure).
1.4 Select Missing Residues to Repair & Prepared Settings
Select Missing Residues to Repair: There are no missing residues in this case, so no selection is needed.
Prepared Settings: Uncheck the Energy Minimization option.
Name the job "SARS-CoV-2 Protein Preparation" and click "Submit" to submit the task.
1.5 Viewing Protein Preparation Results
Protein Preparation calculations typically complete within a few seconds to minutes. Once completed, access the job by clicking Job.
In the Job List, click the "show" button next to the "SARS-CoV-2 Protein Preparation" task to display the prepared protein structure in the 3D Workspace and check the protein structure.
2. MM PB/GBSA Structure
2.1 Accessing MM PB/GBSA Structure
Navigate to MM PB/GBSA Structure through the general menu bar on the left: Function → Binding Affinity Evolution → MM PB/GBSA Structure.
2.2 Select a Prepared Protein
Click "Project," then select "SARS-CoV-2 Protein Preparation" as the prepared protein structure in the pop-up window, and click OK.
After clicking OK, the system automatically checks if the input protein meets the computational requirements, showing the status "Processing";
In less than a minute, the system will mark the protein as "Valid." Click "Next."
Note: MM PB/GBSA supports a maximum of 2000 atoms and a molecular weight limit of 12800 for the protein. Non-standard amino acids, co-crystallized small molecules, and other solvents are not permitted. For operating instructions on more protein selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
2.3 Select Ligands
Click "From File" and select the compound structures 7O46_17.sdf, 7NEO_15.sdf, 7B77_8.sdf, 7B5Z_6.sdf, 7B2J_5.sdf from the local folder to import the ligand structures.
The five ligand structures will be loaded into "Selected File". The system automatically checks if the input Ligand meets the computational requirements, showing the status "Processing," and then marks the ligand as "Valid." Click Next.
Note: For operating instructions on more ligand selection methods, please refer to "Protein Selection, Ligand Selection and Pocket Setting".
2.4 Confirm and Setting
In this tutorial, select "PBSA" for Solvation Mode.
Check the "Total Decomposition Contribution Analysis" option to decompose residue energy.
Name the job "SARS-CoV-2 MMPBSA."
Keep the remaining parameters at their default values and click "Submit" to submit the task.
| Parameter | Option | Meaning | Note |
| Solvation Mode (Determine the surface area generation method) | PBSA | The PBSA method is based on the Poisson-Boltzmann equation, which describes the electrostatic potential of charged particles in a continuous dielectric medium | The PBSA method typically offers greater accuracy than GBSA, especially for systems involving complex ion distributions |
| GBSA | The GBSA method is based on the Generalized Born (GB) approximation, a simplified version of the Poisson-Boltzmann equation, used to estimate electrostatic effects during solvation | GBSA is computationally more straightforward and faster than PBSA, often used for large-scale screening or prediction tasks | |
| Protein Force Field | Amber03、Amber99sb、Amber99sb-ildn、Amber99sb-star-ildn-mut | Selective protein force field | Recommend using Amber03 |
| Ligand Force Field | gaff2、gaff | Selective match field | Recommend using gaff2 |
| Energy Minimization | Choose whether to minimize energy | ||
| Dielectric Constant | The dielectric constant is a physical quantity that describes how an electric field (the force between charges) is diminished in a material. | Recommended default values. | |
| Implicit Solvent Dielectric Constant | The implicit solvent dielectric constant represents the dielectric constant of the continuous dielectric field in the implicit solvent model | Recommended default values | |
| Non-polar Surface Constant | Non-polar surface constants usually refer to parameters used to calculate the free energy of non-polar surfaces | Recommended default values. The value of the nonpolar surface constant should be calibrated using experimental data or obtained from the literature | |
| Total Decomposition Contribution Analysis | Calculate the energy contribution of amino acid residues |
3. MM PB/GBSA Results Analysis
3.1 Accessing Task Results
Navigate to Job → Job List in the left general menu bar, find the "SARS-CoV-2 MMPBSA" computation task, and click "Show."
3.2 Results List
The calculated binding free energy results are displayed in the following table. By comparing with experimental IC50 value rankings, the predicted binding energy rankings of the five compound inhibitors are quite accurate.
The results indicate that the Uni-MM PB/GBSA module can accurately predict the ranking of binding free energies of compound inhibitors. It can be used to predict the binding affinity of compound inhibitors to proteins, providing theoretical support for drug design and optimization processes.
| PDB ID | Compound | IC50 (μM) | MM PBSA(kcal/mol) |
| 7O46 | 17 | 0.15 | -14.559 |
| 7NEO | 15 | 6.60 | -14.521 |
| 7B2J | 5 | 7.20 | -11.179 |
| 7B5Z | 6 | 38.50 | -7.074 |
| 7B77 | 8 | 79.30 | -6.625 |
MM PB/GBSA calculation results
-
ΔG are provided in 3 forms: solvent, gas, and sum:
-
3.3 Pose Display
Click the "3D" button to examine the protein and ligand conformations in the 3D Workspace.
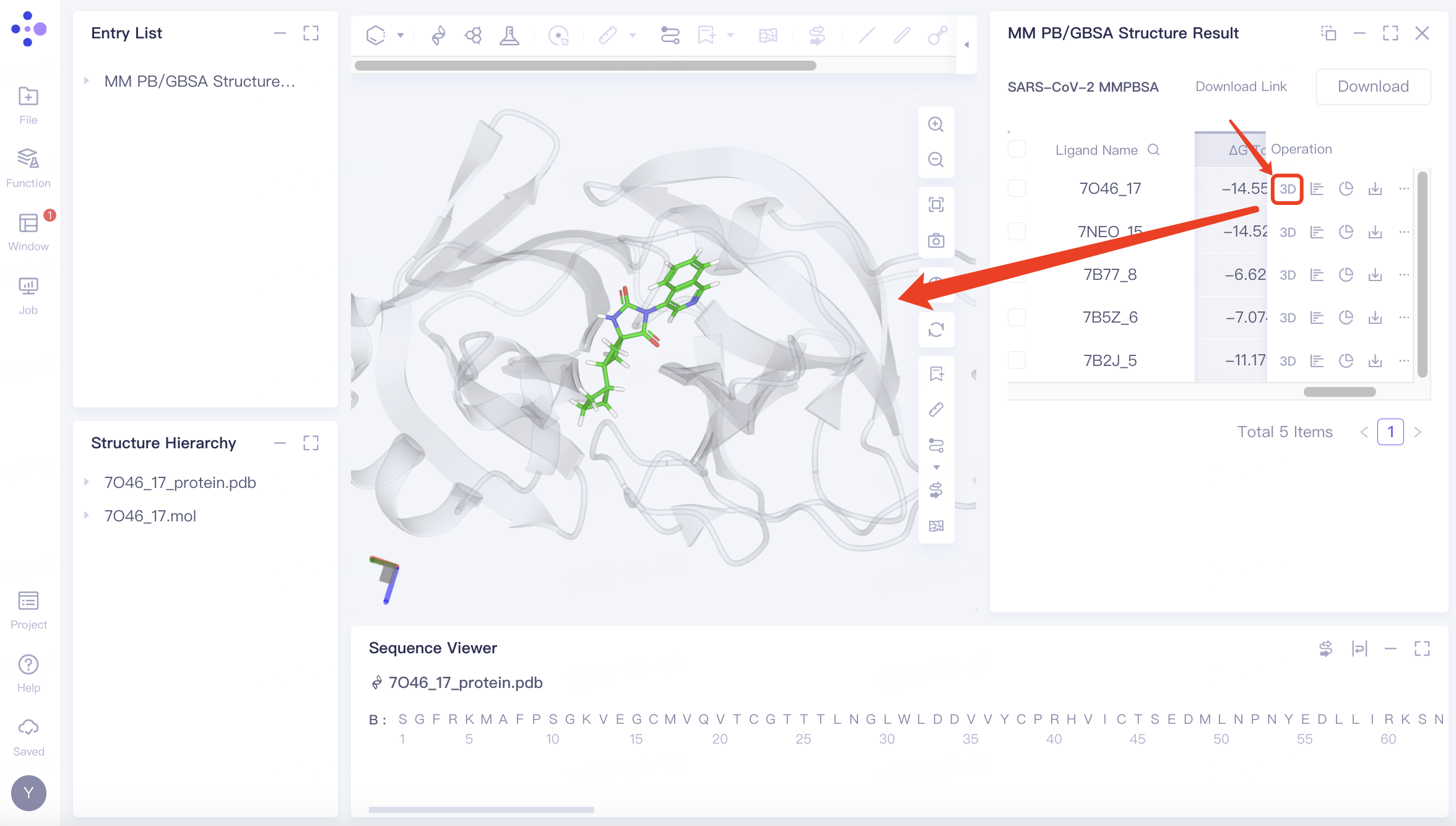
Related operations:
| Button | Meaning | Interface display | Note |
ΔG | Review the numerical values of the various energy components contributing to the ΔG (binding free energy) | 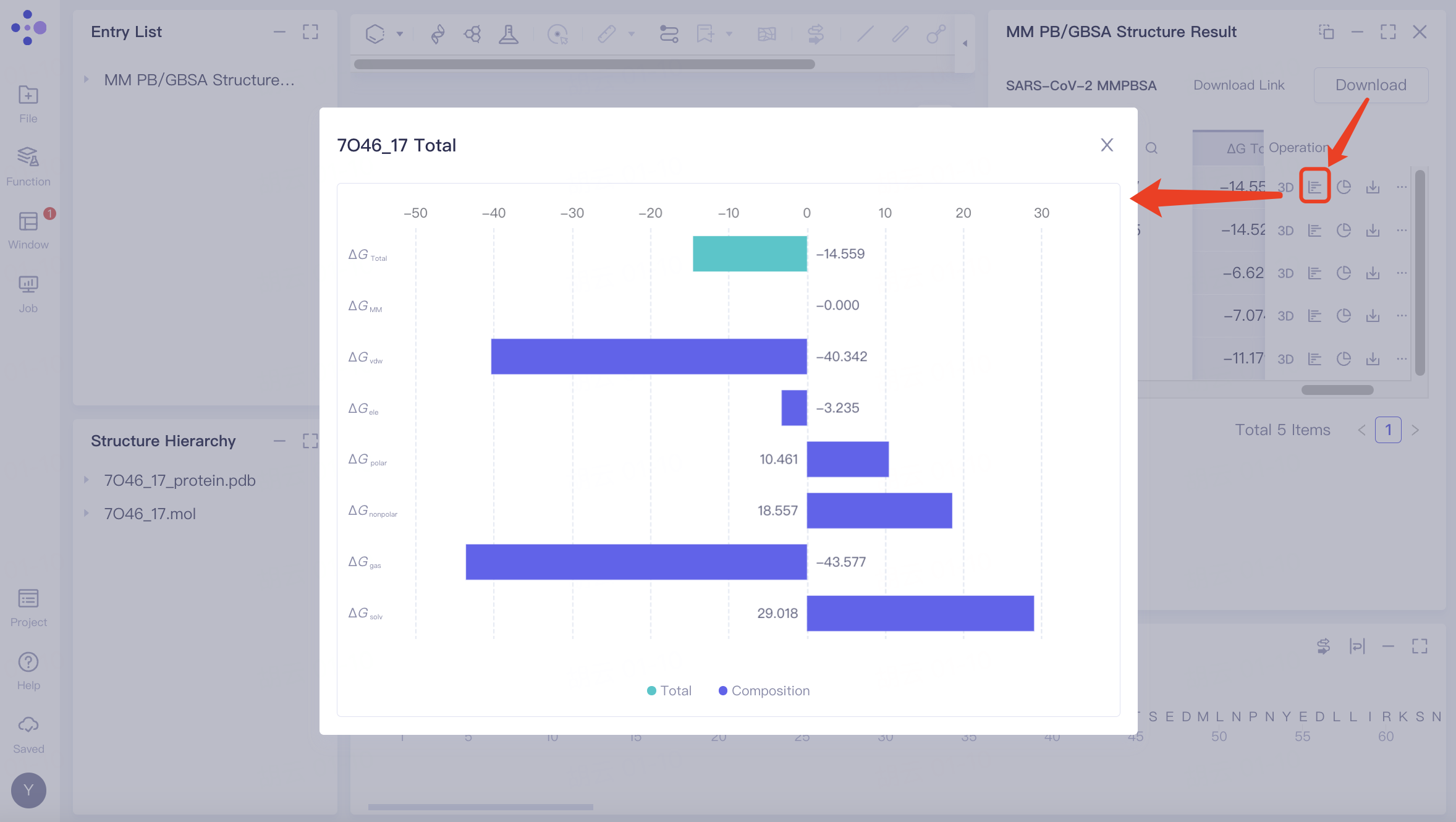 | |
Decomposition | Amino acid residue energy decomposition | 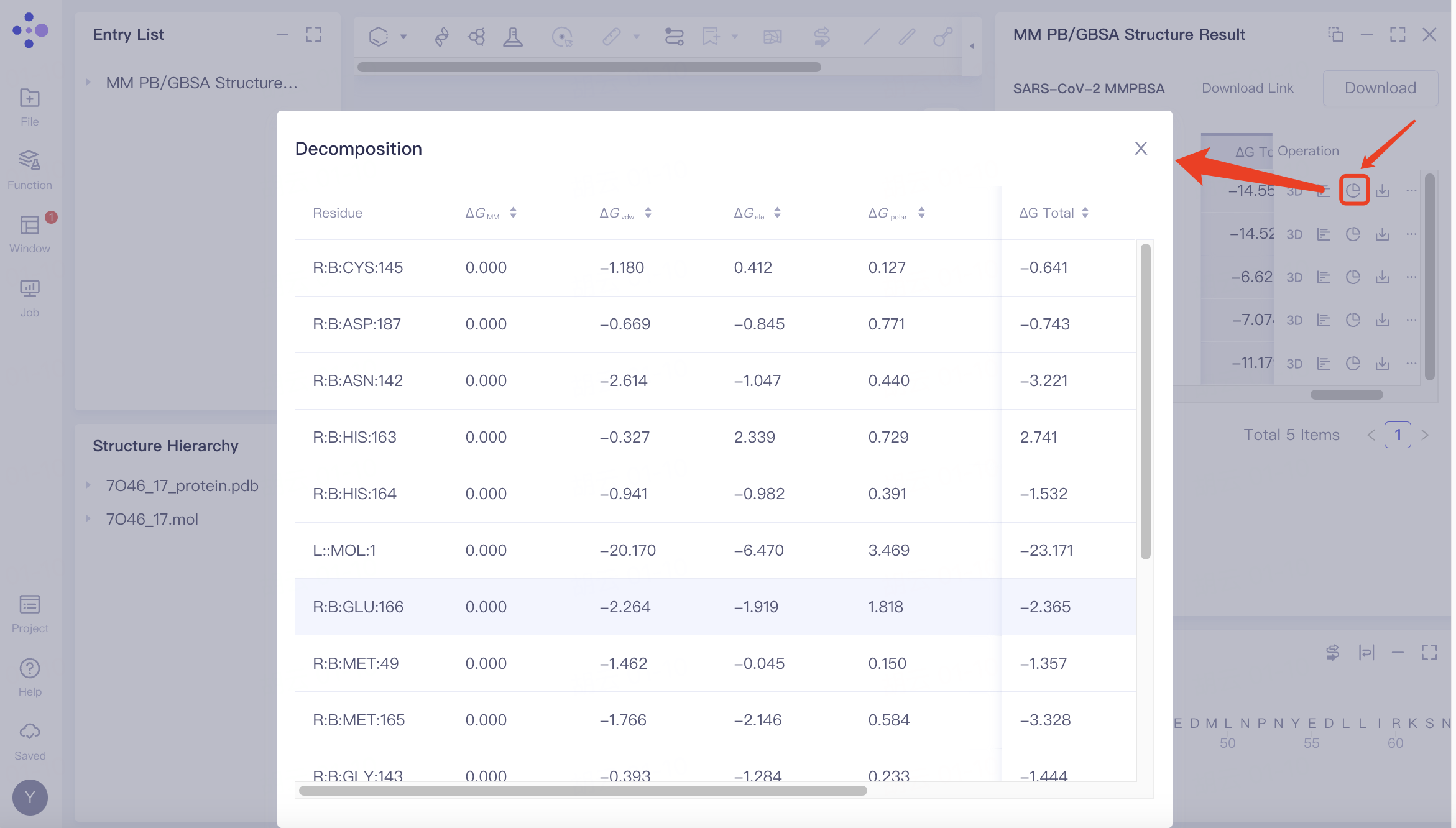 | If "Total Decomposition Contribution Analysis" was selected during the calculation, the detailed breakdown of energy contributions by amino acid residues can be viewed. |
Download | Single download | 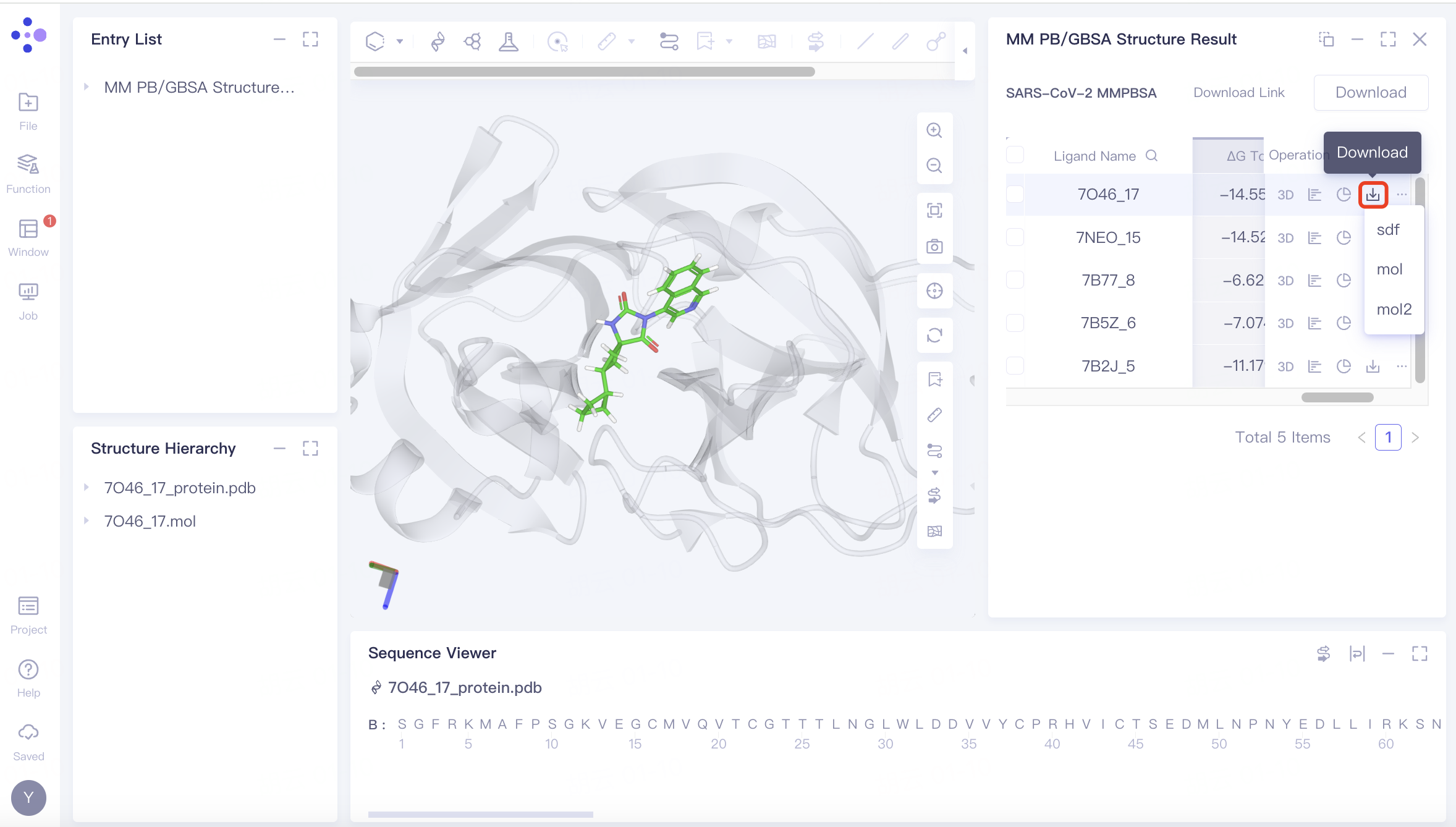 | |
 | Batch download | 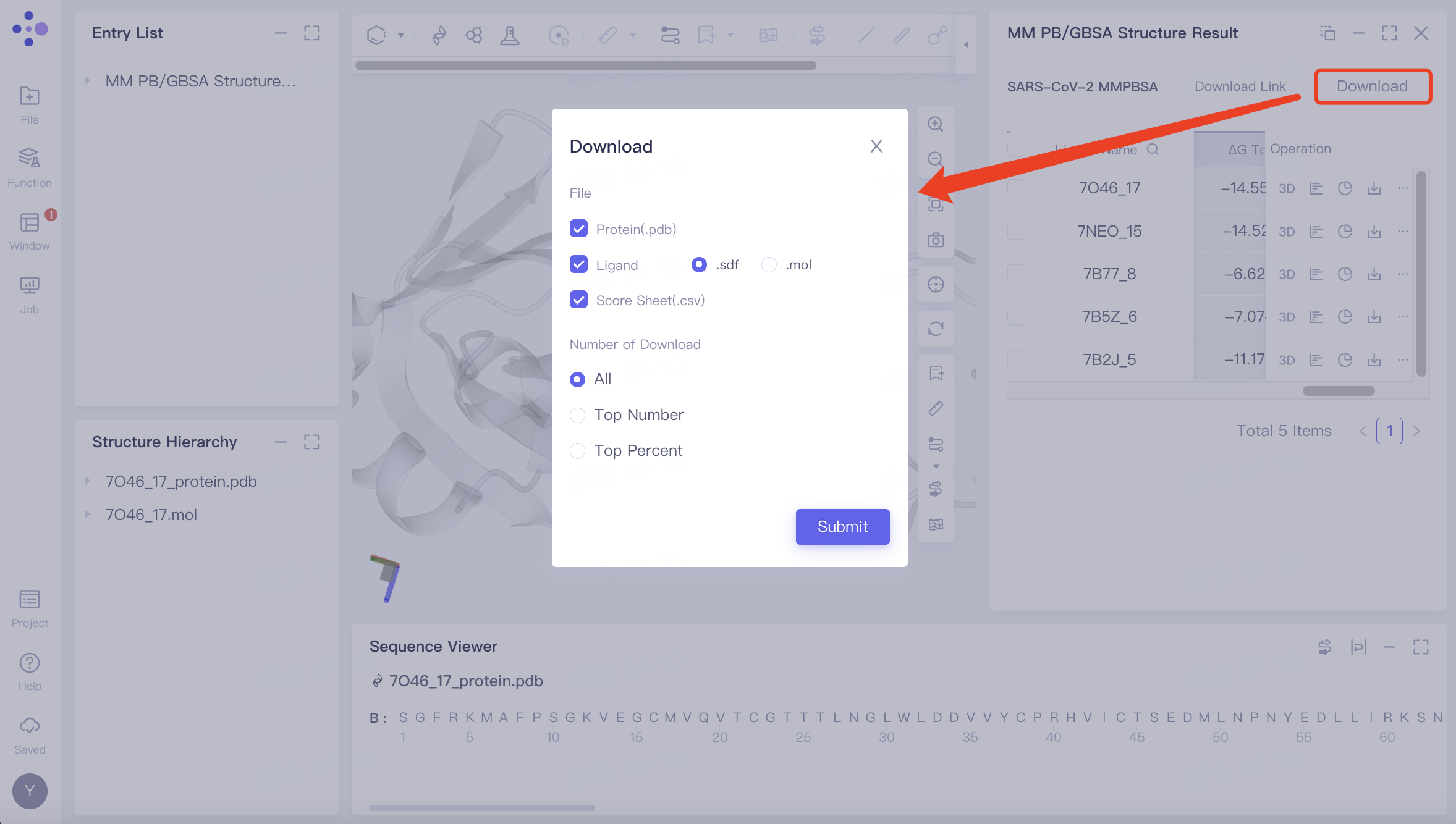 | Download steps: Click the "Download" button and select the parameters you wish to download. A message stating "Link is preparing" will appear; wait for the loading to complete. Once the loading is finished, click the "Download Link" button and use the provided URL link to download the calculation results. |
4. References
[1] Maohua Yang, Zonghua Bo, Tao Xu, Bo Xu, Dongdong Wang, Hang Zheng, Uni-GBSA: an open-source and web-based automatic workflow to perform MM/GB(PB)SA calculations for virtual screening, Briefings in Bioinformatics, Volume 24, Issue 4, July 2023, bbad218, https://doi.org/10.1093/bib/bbad218
[2] Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 7, 2905–2920