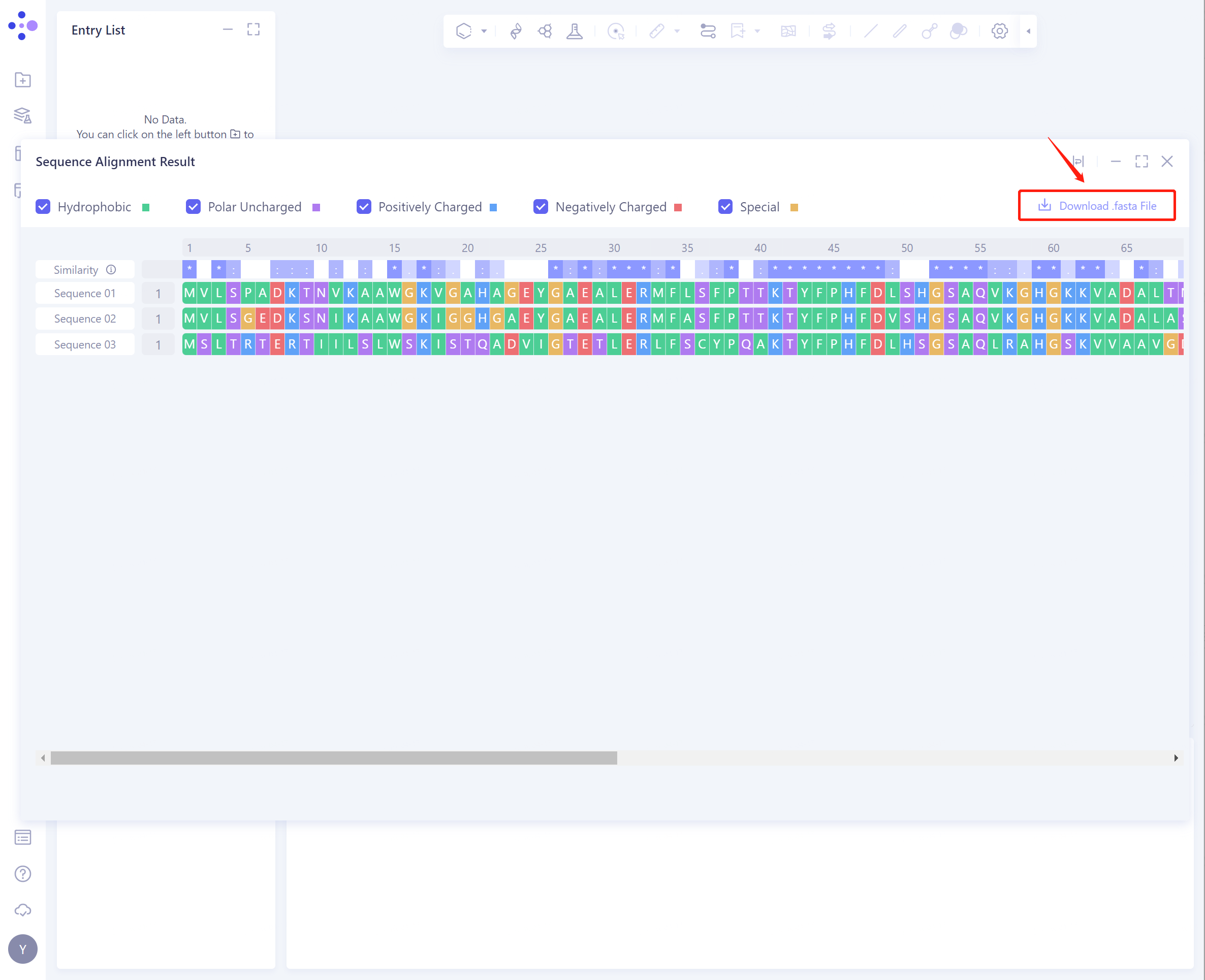
Sequence Alignment
Introduction
Sequence alignment is a method to compare two or more biological sequences (protein sequences, nucleotide sequences), which can help us understand the similarity between these sequences and how they change in the process of evolution.
In this tutorial, you will learn to perform multiple sequence alignment using the Sequence Alignment module of the Hermite ® platform. The examples in this tutorial are from the Hemoglobin subunit sequences of human, mouse and goat. By comparing their sequences, we can analyze the conserved sites of hemoglobin gene in the process of biological evolution, the evolutionary conservation of different amino acid types, and the overall sequence similarity.
Note: Currently, this function only supports the alignment of amino acid sequences.
Input sequence used in this tutorial:
sp |P69905| HBA_HUMAN Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTL AAHLPAEFTPAVHASLDKFLASVSTVLTSKYR|
sp |P01942| HBA_MOUSE Hemoglobin subunit alpha OS=Mus musculus GN=Hba PE=1 SV=2
MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHGKKVADALASAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTL ASHHPADFTPAVHASLDKFLASVSTVLTSKYR|
sp |P13786| HBAZ_CAPHI Hemoglobin subunit zeta OS=Capra hircus GN=HBZ1 PE=3 SV=2
MSLTRTERTIILSLWSKISTQADVIGTETLERLFSCYPQAKTYFPHFDLHSGSAQLRAHGSKVVAAVGDAVKSIDNVTSALSKLSELHAYVLRVDPVNFKFLSHCLLVTL ASHFPADFTADAHAAWDKFLSIVSGVLTEKYR|
Input sequence files used in this tutorial:
1. Create a task
1.1 Entrance
- The left general menu bar is Menu → Function → Biomacromolecule → Sequence Alignment.
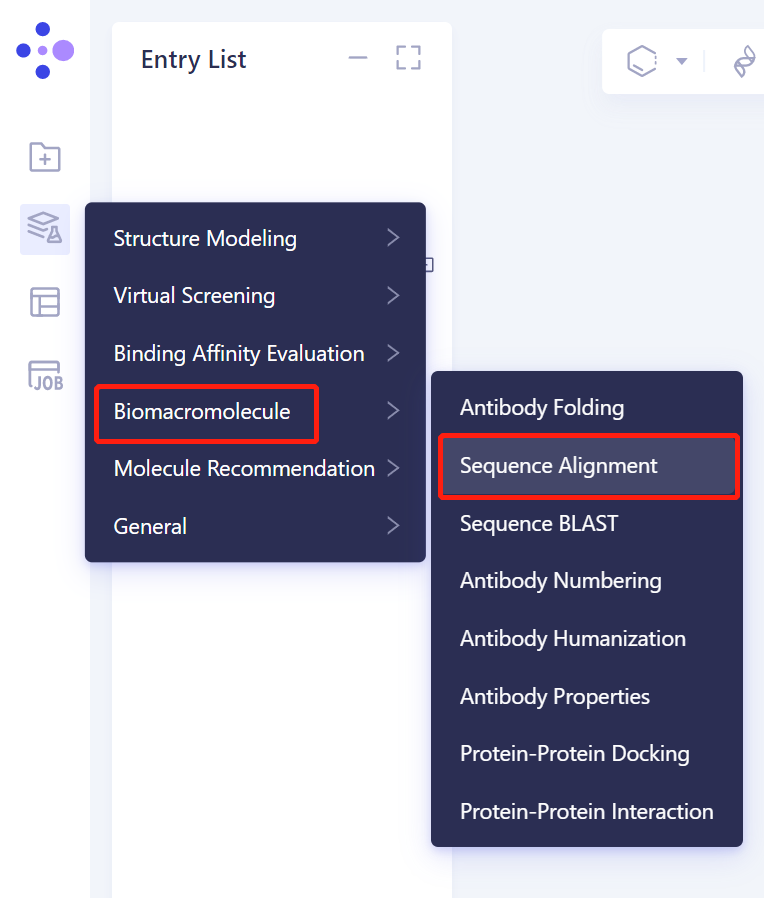
- The operation box of Sequence Alignment appears on the right side (shown in the red box), and the overall interface is as follows:
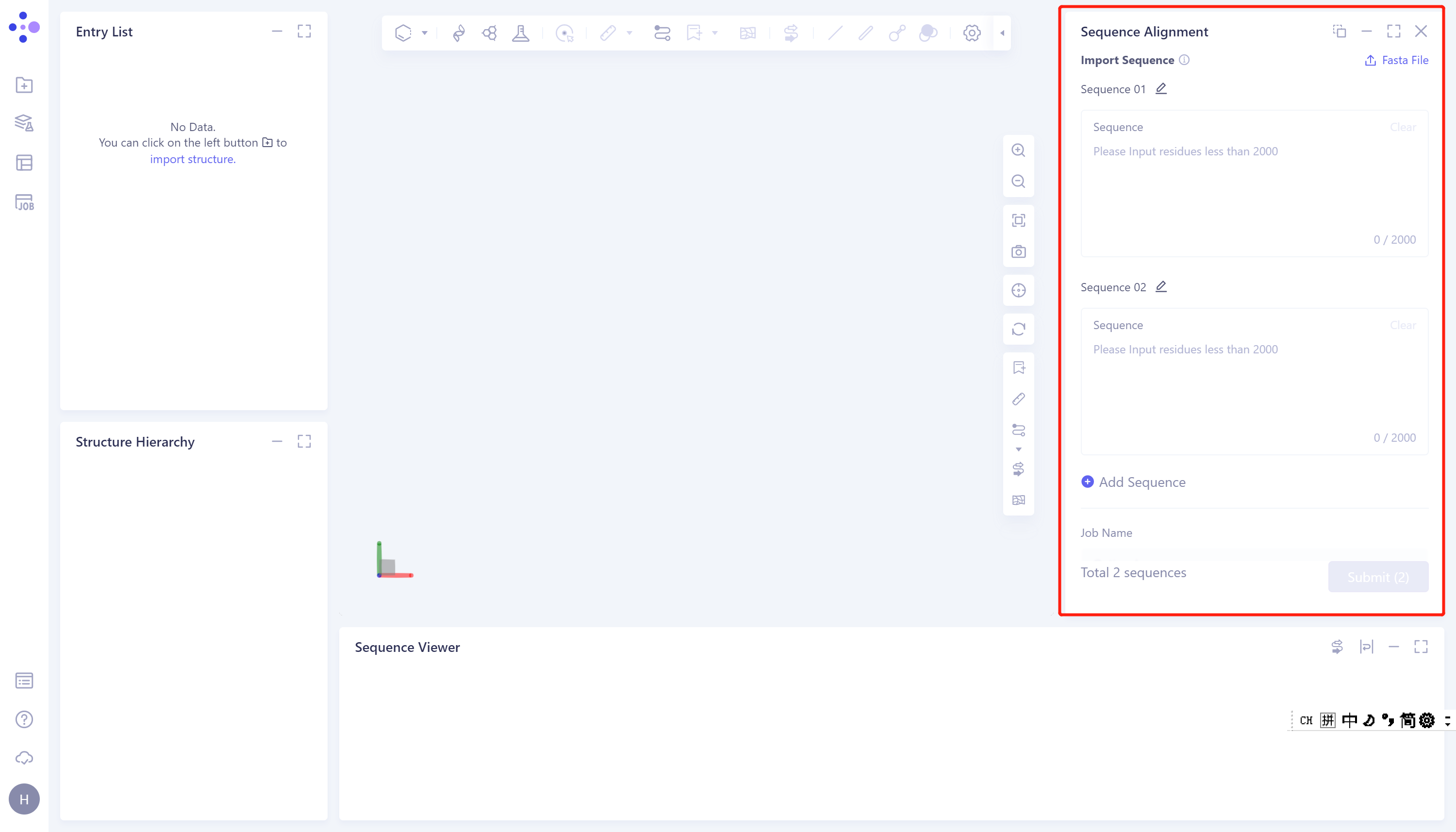
1.2 Operation
- Import Sequence: Input the sequence. You can choose to input the sequence directly or upload the sequence file. In this tutorial, you can choose to input the sequence directly. The input result is shown in the figure below:
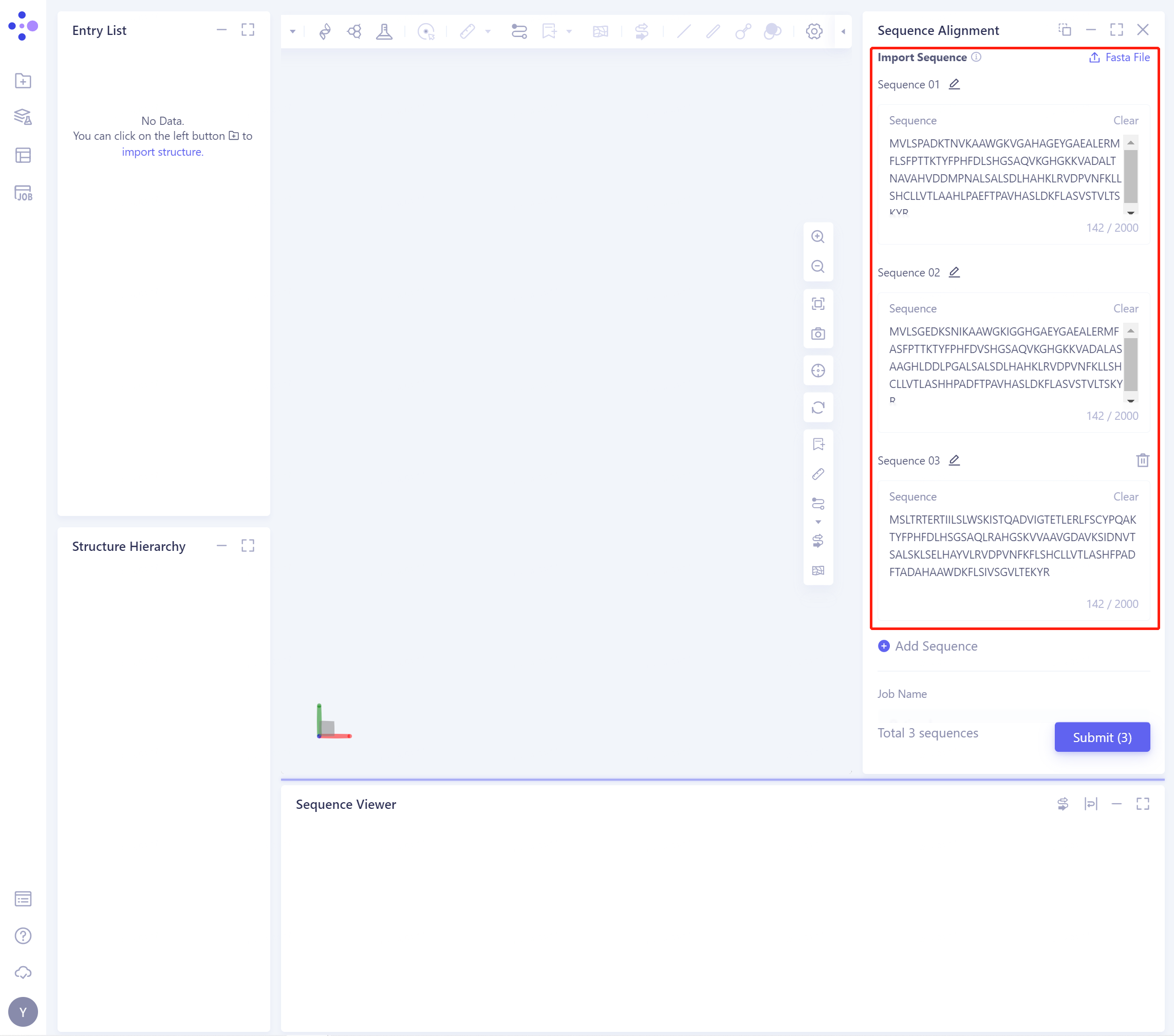
-
Name the task at Job Name: Sequence Alignment.
-
Click Submit to submit the task.
2. Analysis of results
2.1 Entrance
- In the general menu bar on the left, Menu Job → Job List, find the task, and click the Show button in the Operation column to display the task results.
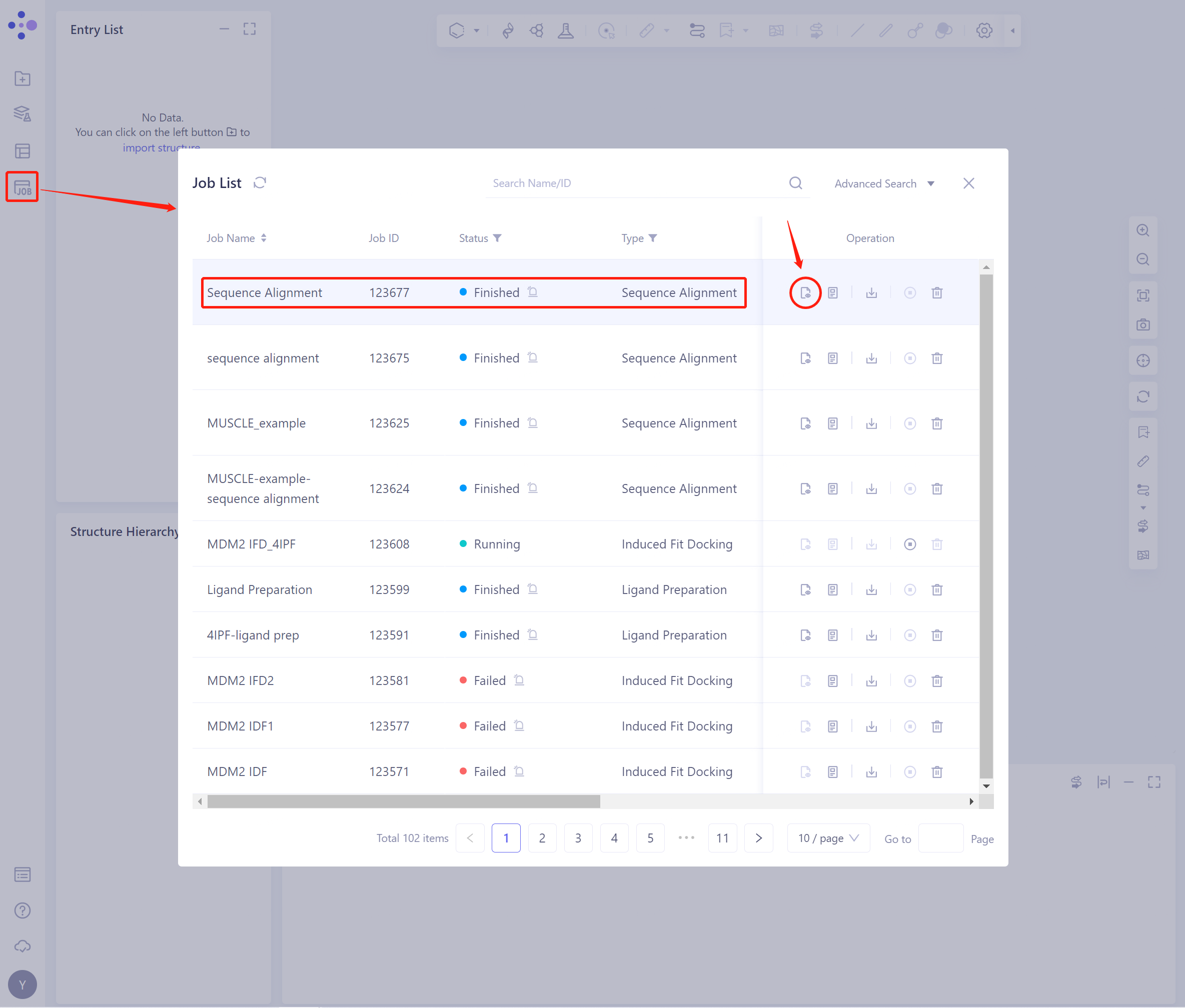
2.2 Operation
2.2.1 Presentation of results
- The result interface is as shown in the figure:
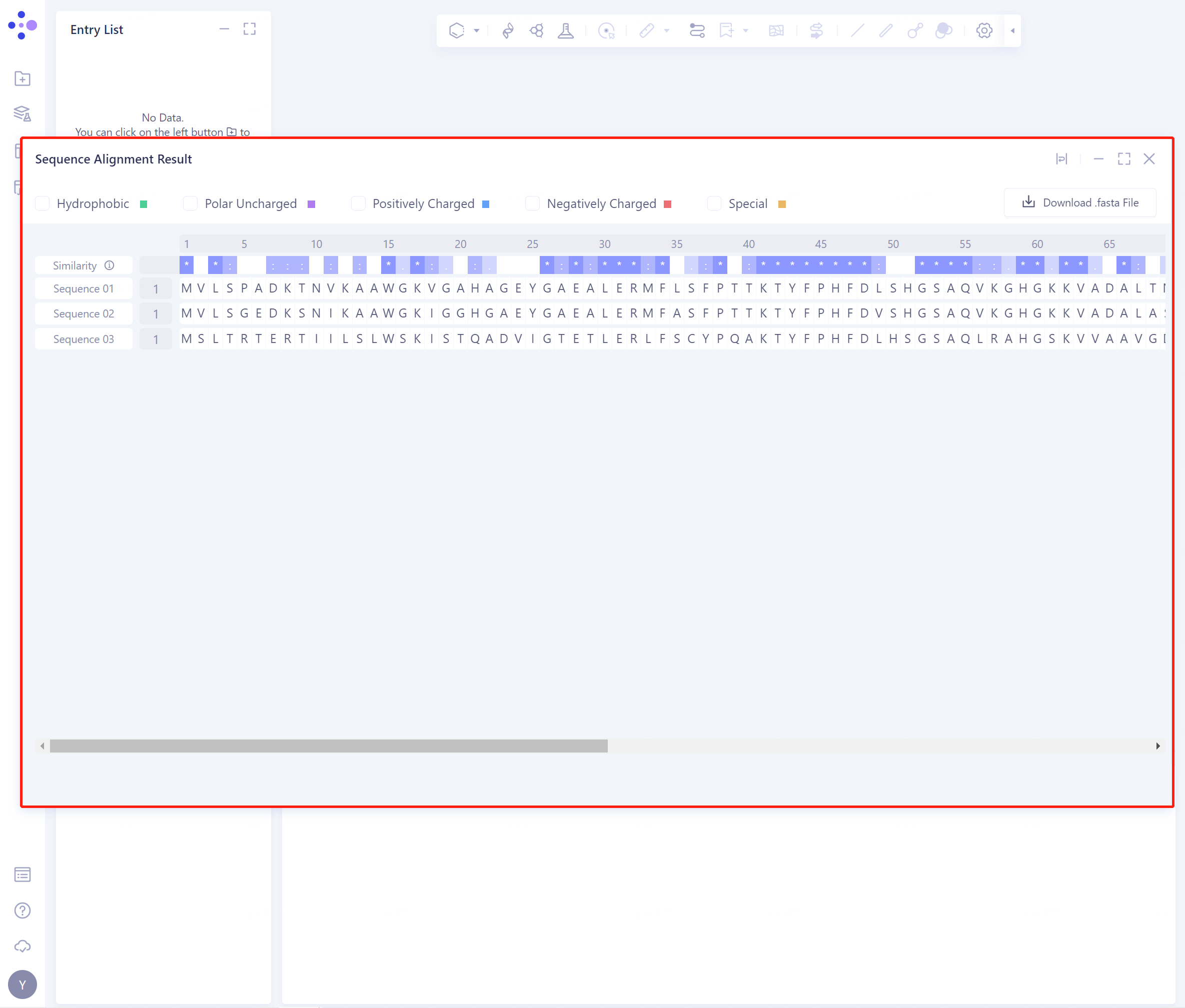
2.2.2 Explanation of results
-
The shade of blue in the Similarity row indicates the degree of conservation of the sequence, which can be used to analyze the evolutionary conservation of different domains.
-
Different types of amino acids can be marked in the sequence by checking Hydrophobic, Polar Uncharged, Positively Charged, Negatively Charged, and Special. It was found that the hydrophobic amino acids in the gene had the highest evolutionary conservation (green marker residues).
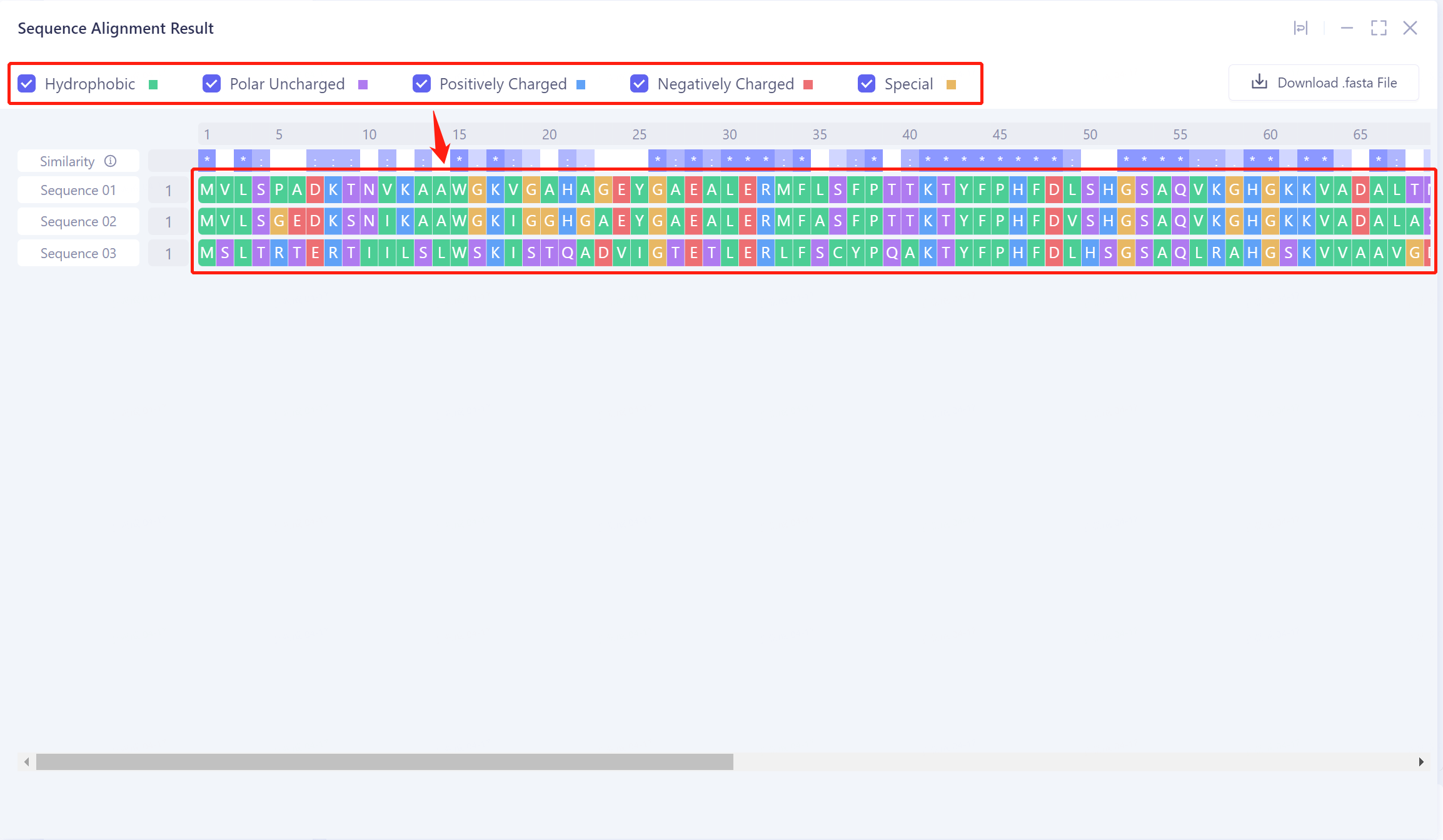
- Click the Download. Fasta File button to download the aligned.fasta file for further analysis.