Protein-Protein Docking
Introduction
Protein docking is a technique to predict the recognition and interaction between proteins. In Hermite, protein-protein Docking can be used to implement protein-protein docking calculations.
SUMOylation is a post-translational covalent linker of SUMO family (SUMO-1, SUMO-2, or SUMO-3) proteins that regulate many cellular pathways. The crystal structure of 5ELU is a protein-protein complex of human small ubiquitin modifier 2 (chain B) and synthetic protein SUMO-Affirmer-S2B3 (chain a), which can inhibit SUMO-1- and SUMO-2/3-mediated protein interactions. This tutorial is based on the Protein-Protein Docking module in the Hermite platform to Redock the protein-protein complex structure in 5ELU.
The data used in this tutorial is as follows:
1. Import Protein Structure File
- Left general menu bar 'File' → 'Import Structure' → Click 'Select Files' to select '5ELU_receptor' and '5ELU_ligand' → Click 'Import' to upload protein structure
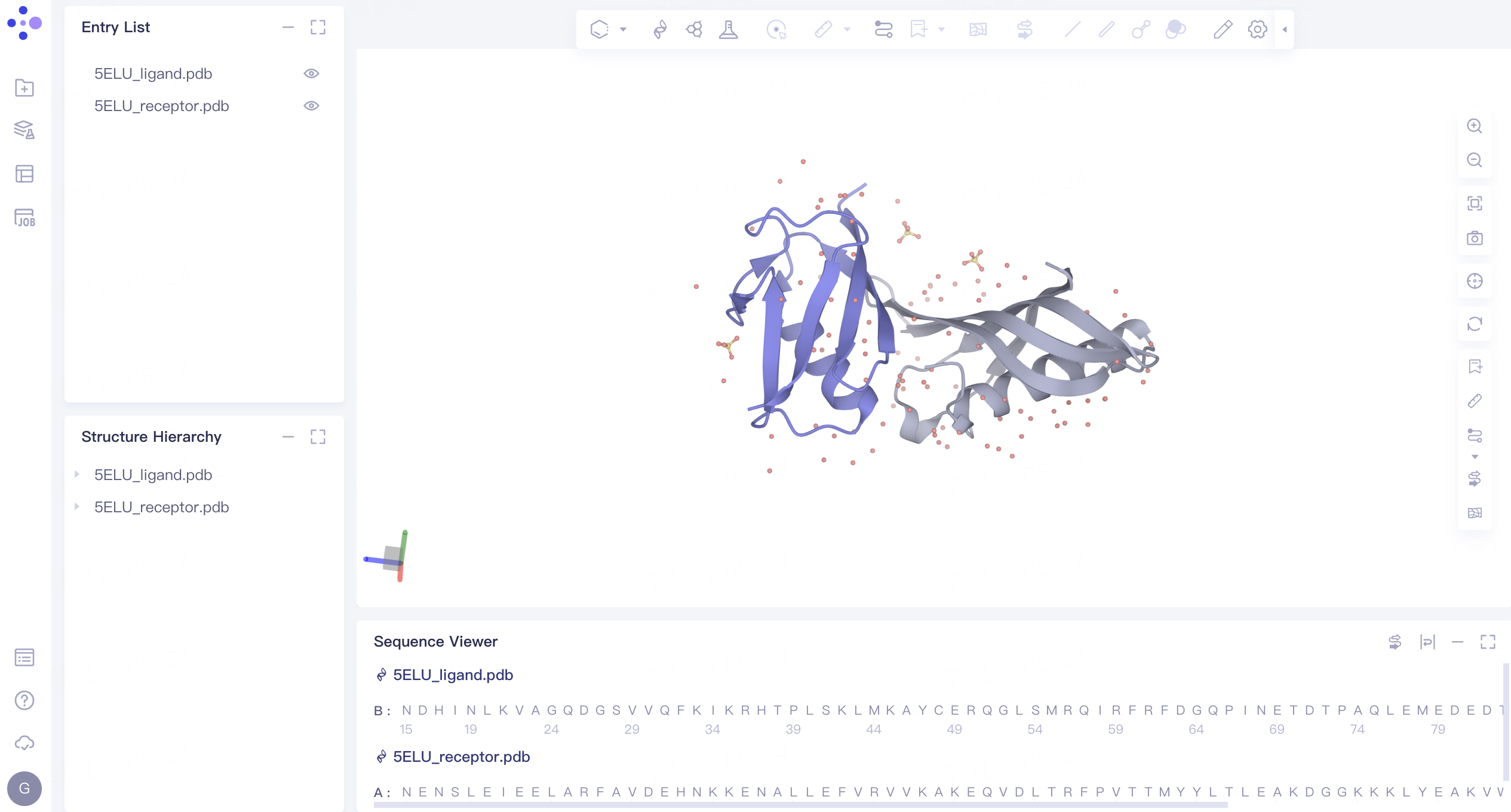
- The protein structure is displayed in the 3D Workspace window as follows:
2. Protein-Protein Docking
2.1 Entrance
- The left general menu bar 'Function' → Biomacromolecule → Protein-Protein Docking.
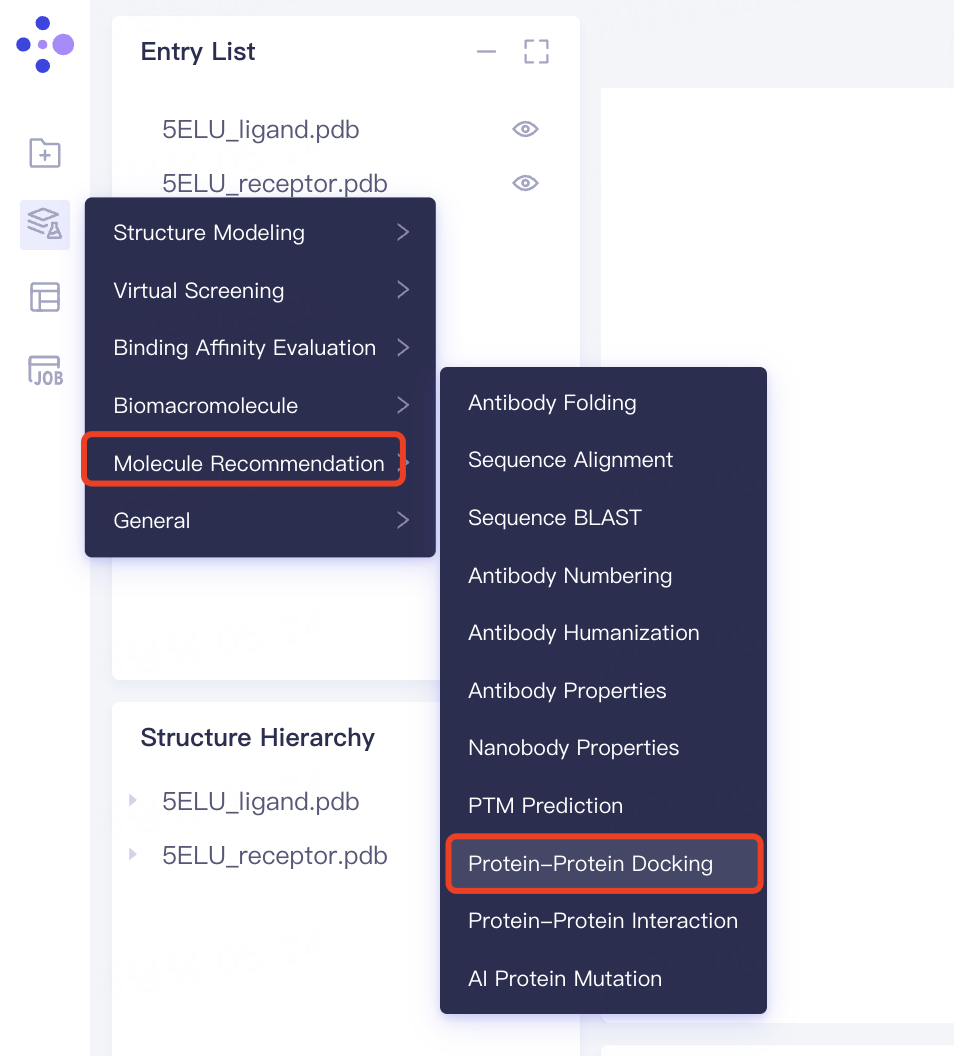
- The interface of 'Protein-Protein Docking' (shown in the red box) appears on the right side, and the overall interface is as follows:
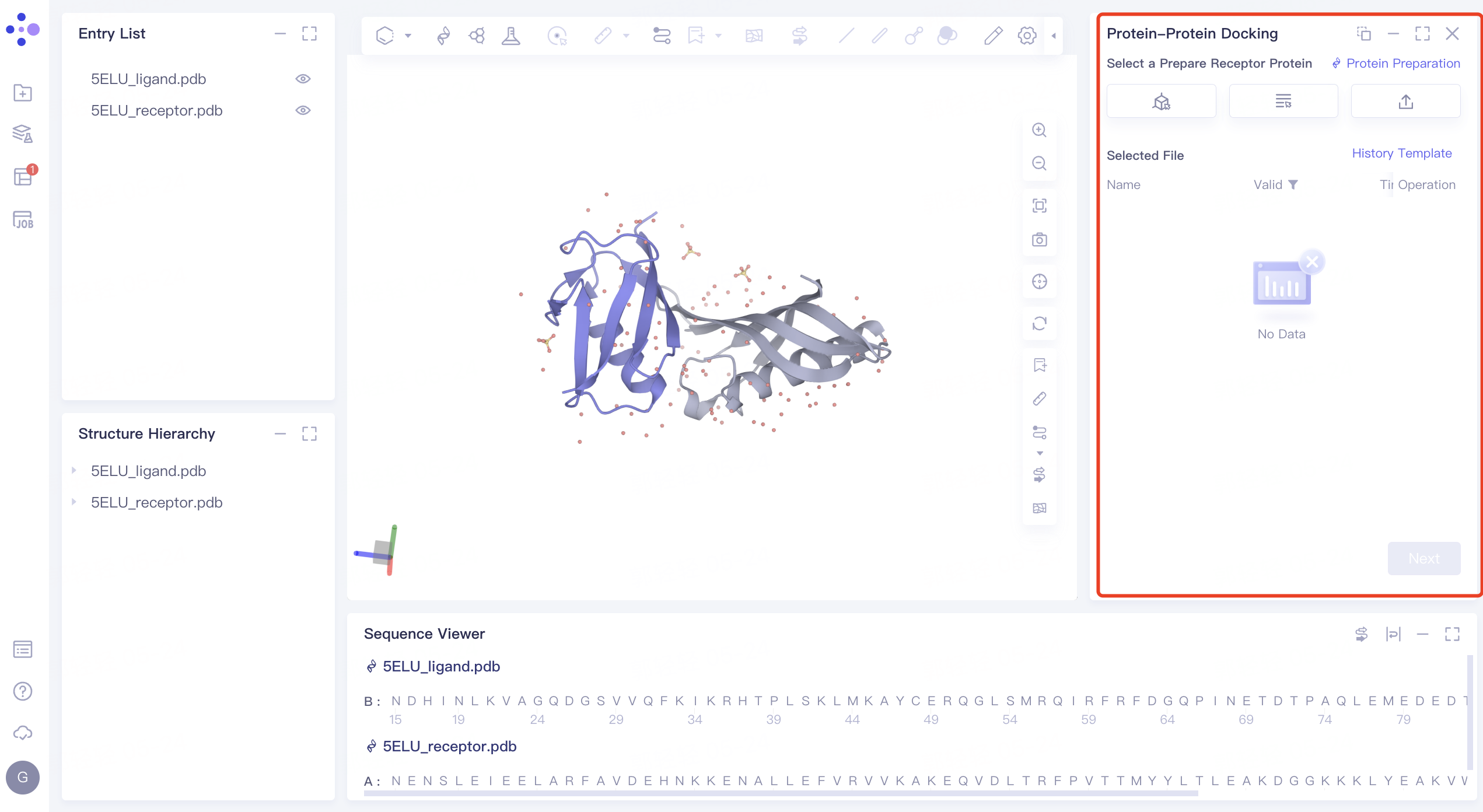
2.2 Receptor Protein File Input and Structure Processing
2.2.1 Receptor Protein File Input
- Select from 3D Workspace: Click the 'Select from 3D Workspace' box → 'Select Structure' interface pops up → Select '5ELU_receptor' in the 'Structure Hierarchy' interface on the left side → The name of the selected protein is displayed in the 'Select Structure' interface. Click 'OK'.
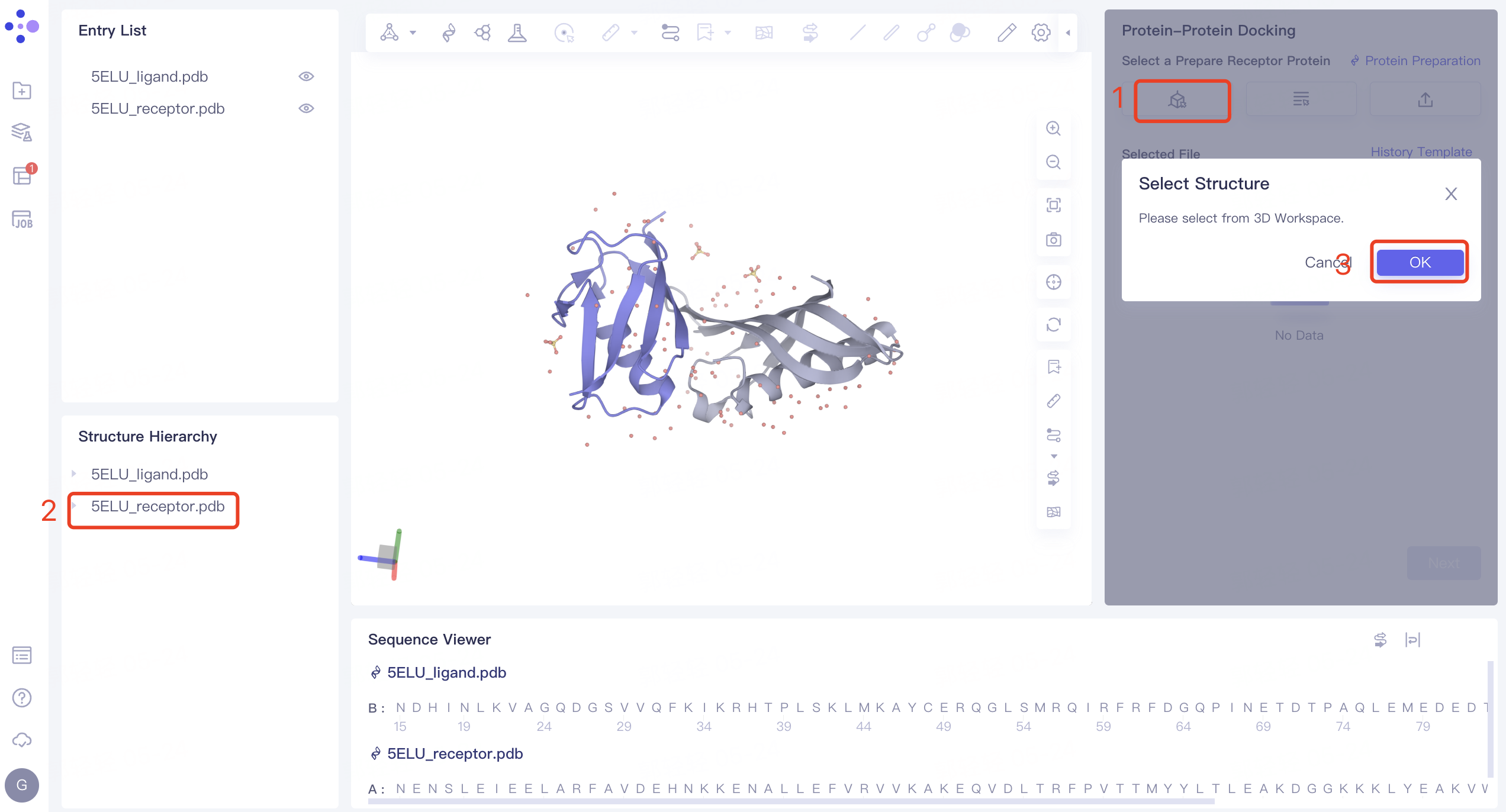
2.2.2 structural treatment of receptor protein
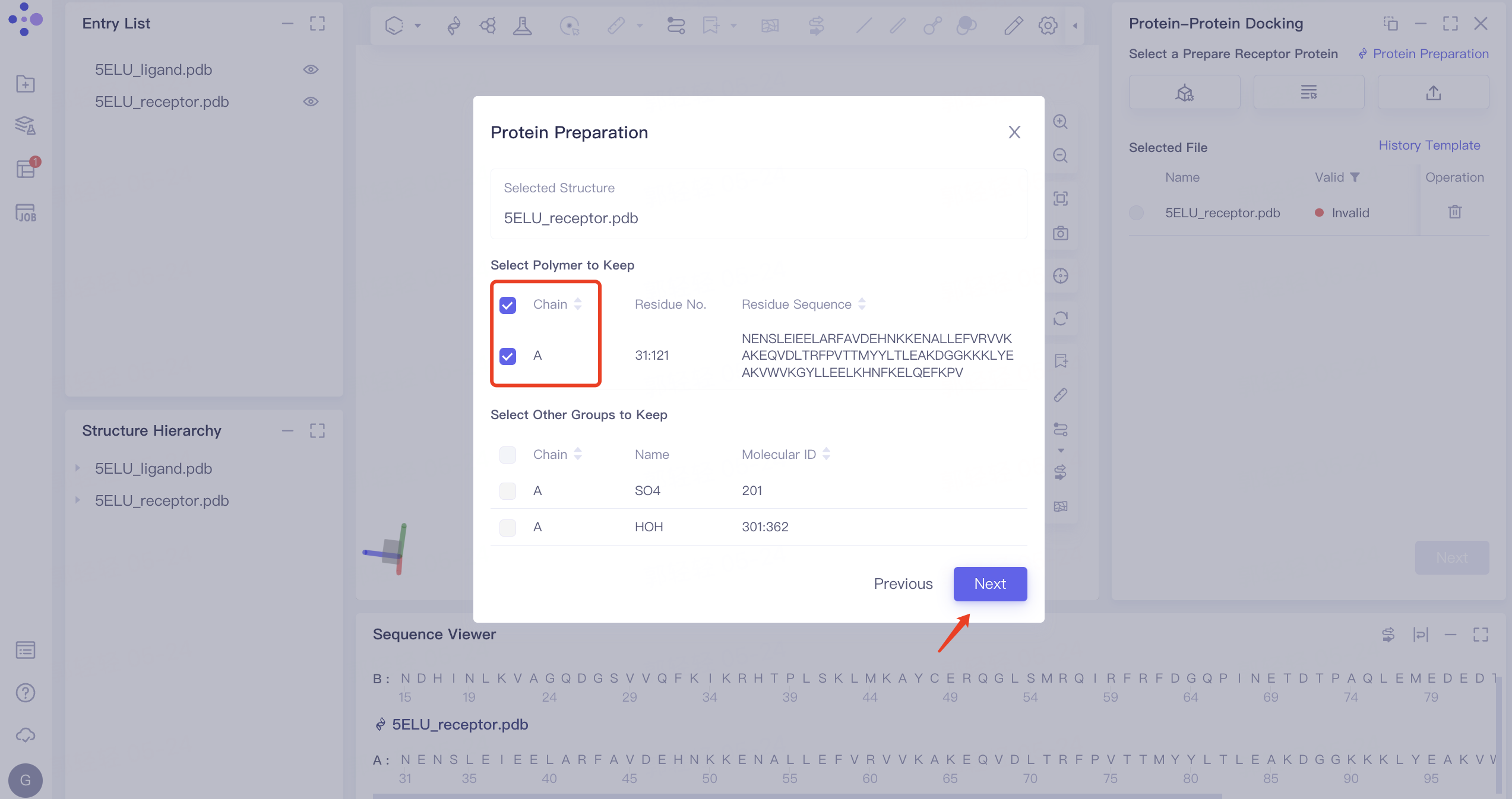
1)'Invalid' displays the status of invalid. In this case, 'Protein Preparation' is required. The protein preparation steps are as follows: click 'Protein Preparation' → the 'Protein Preparation' interface pops up in the middle of the interface → select the receptor structure to be processed → click 'Next'.
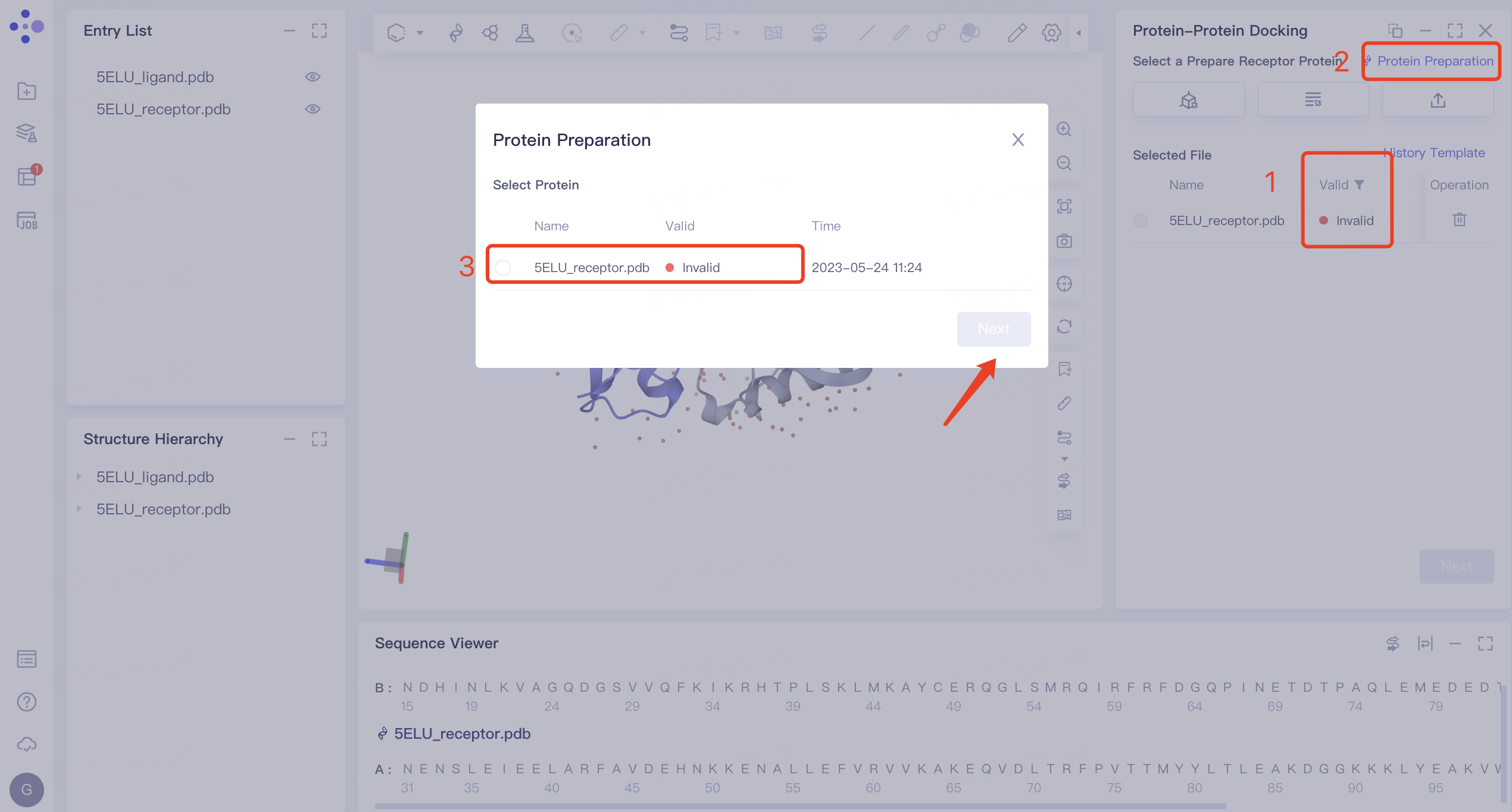
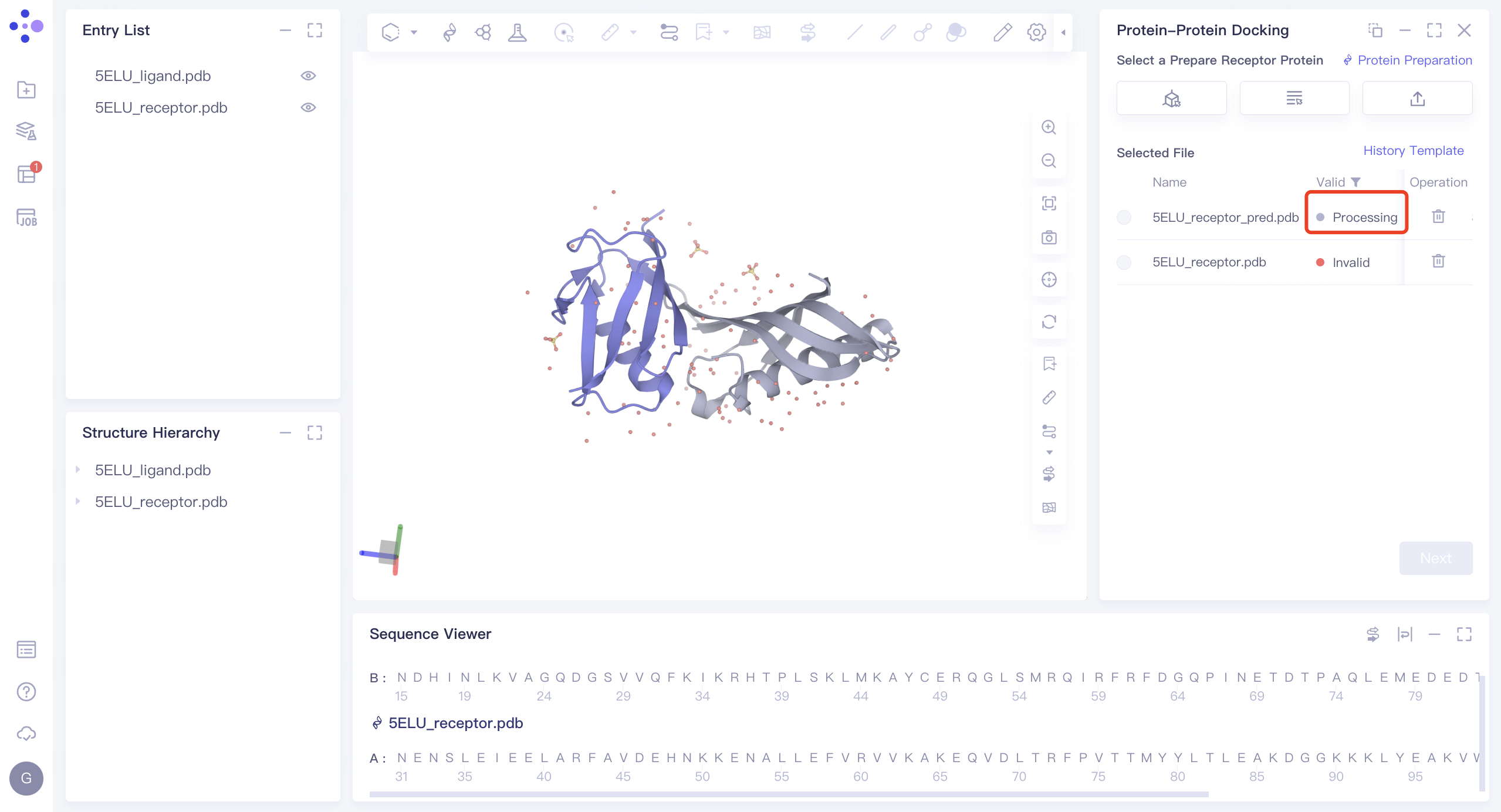
2) Parameter setting: select the receptor protein (Chain A), click 'Next' → prepare the protein according to the default parameters, and click 'Submit' to submit the task. Automatically return to the 'Protein-Protein Docking' window, and the '5ELU_pred.pdb' displays the 'Processing' status. Select the structure after it changes to the 'Valid' status, and click 'Next' to enter the next step.
 | 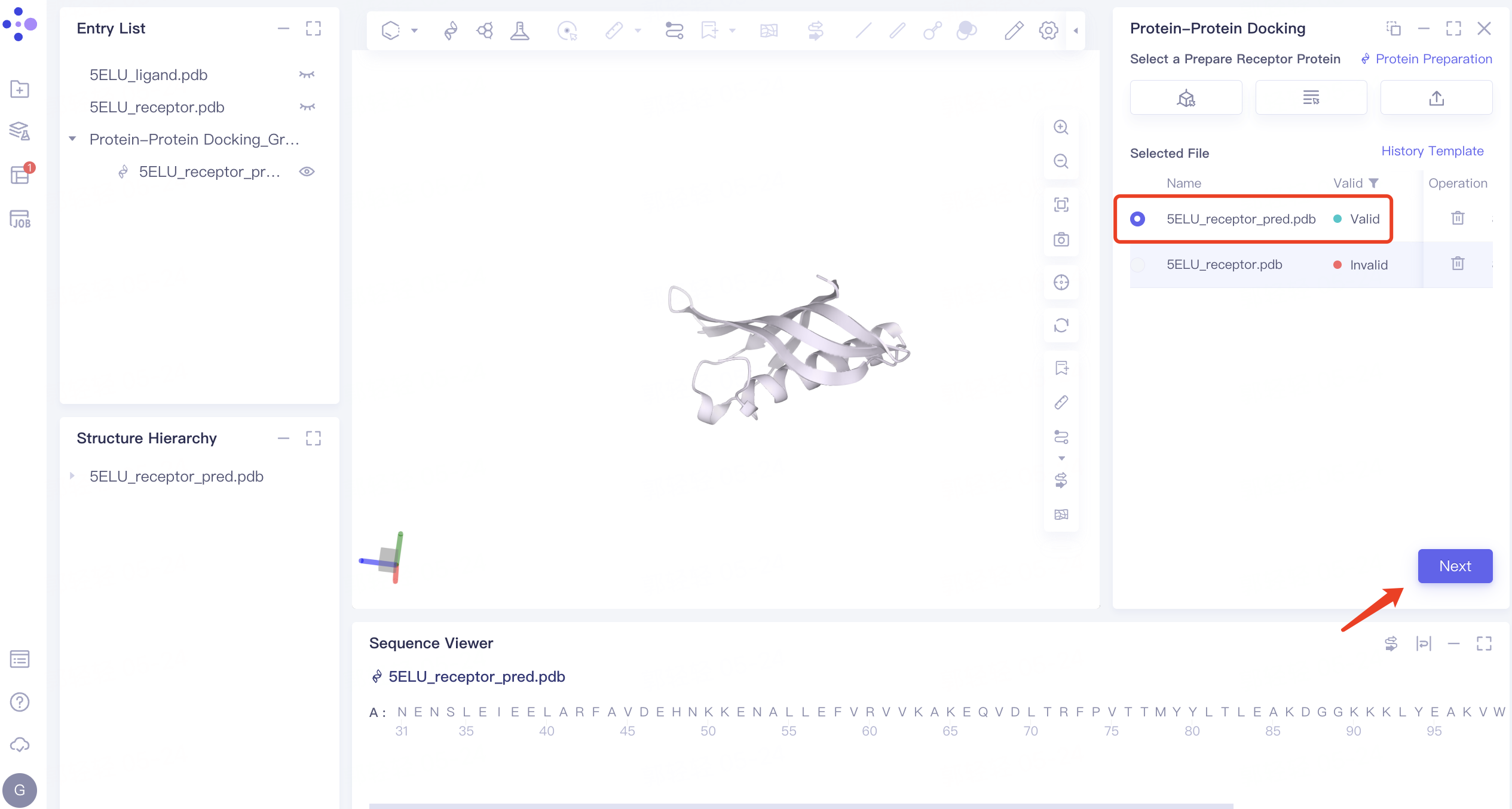 |
2.3 Ligand protein type determination, import, and processing
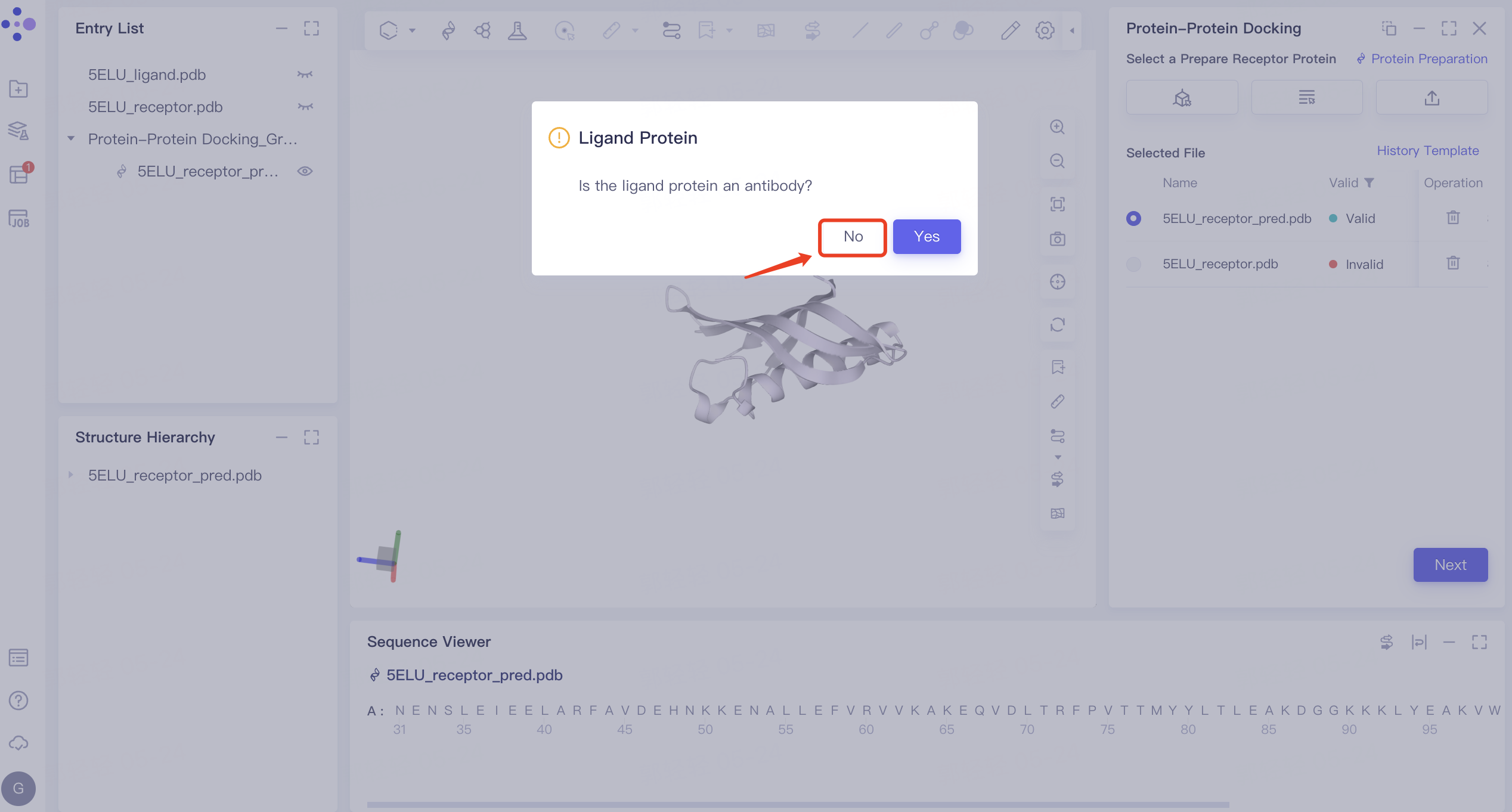
2.3.1 Ligand Protein Type Determination
- The 'Ligand Protein' interface pops up, asking to confirm whether the ligand protein is an antibody. Click 'No' here.
2.3.2 Input of Ligand Protein Structure
-
Display '5ELU_ligand' in the 3D Workspace in the Entry List.
- Click the 'Select from 3D' box → 'Select Ligands' interface pops up → Select '5ELU_ligand' in the Structure Hierarchy interface on the left side of the interface → Display the selected ligand name in the 'Select Structure' interface, and click 'OK'.
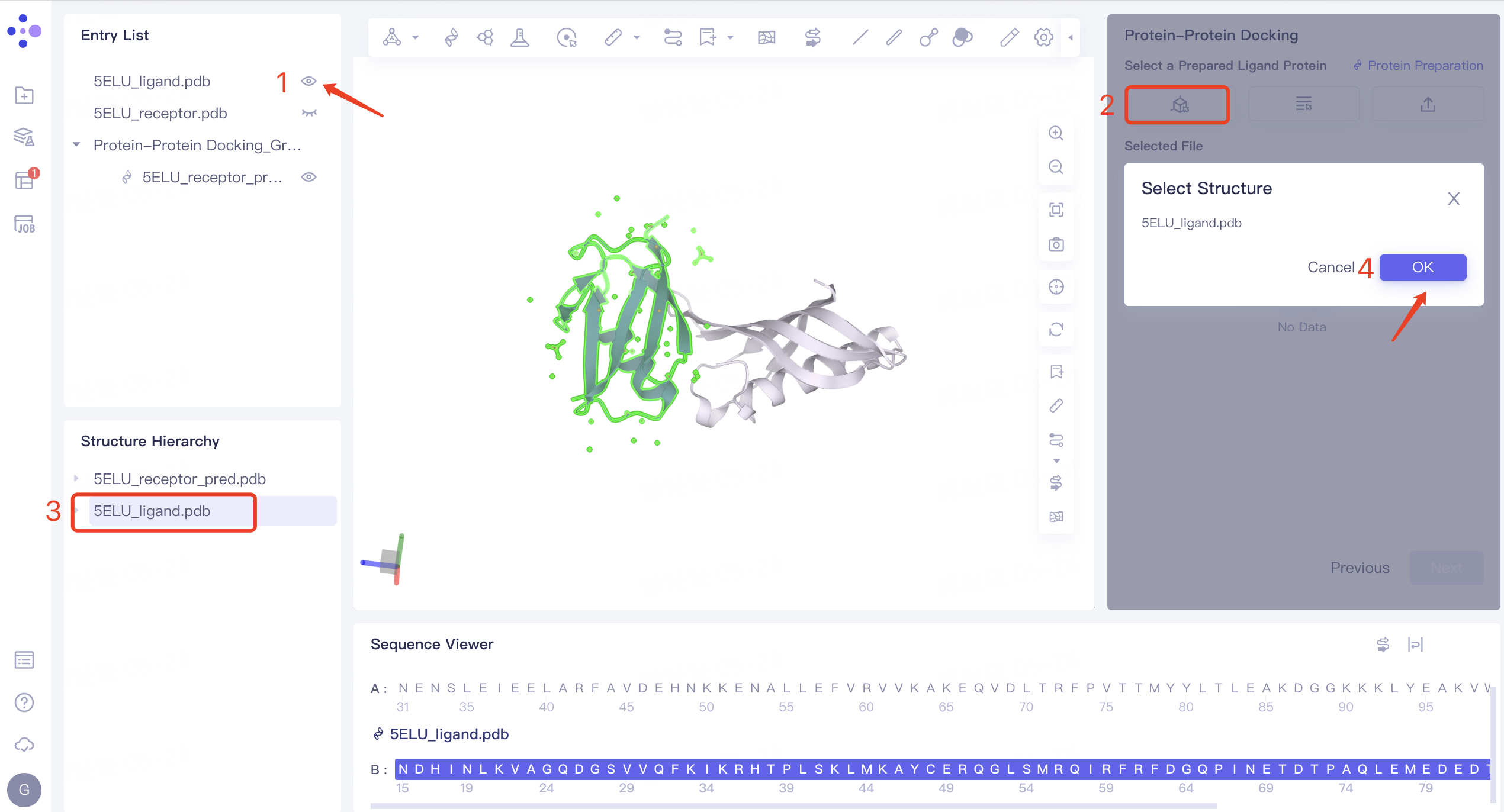
2.3.3 Ligand Protein Structure Treatment
- Same as receptor structure treatment in 2.3.1.
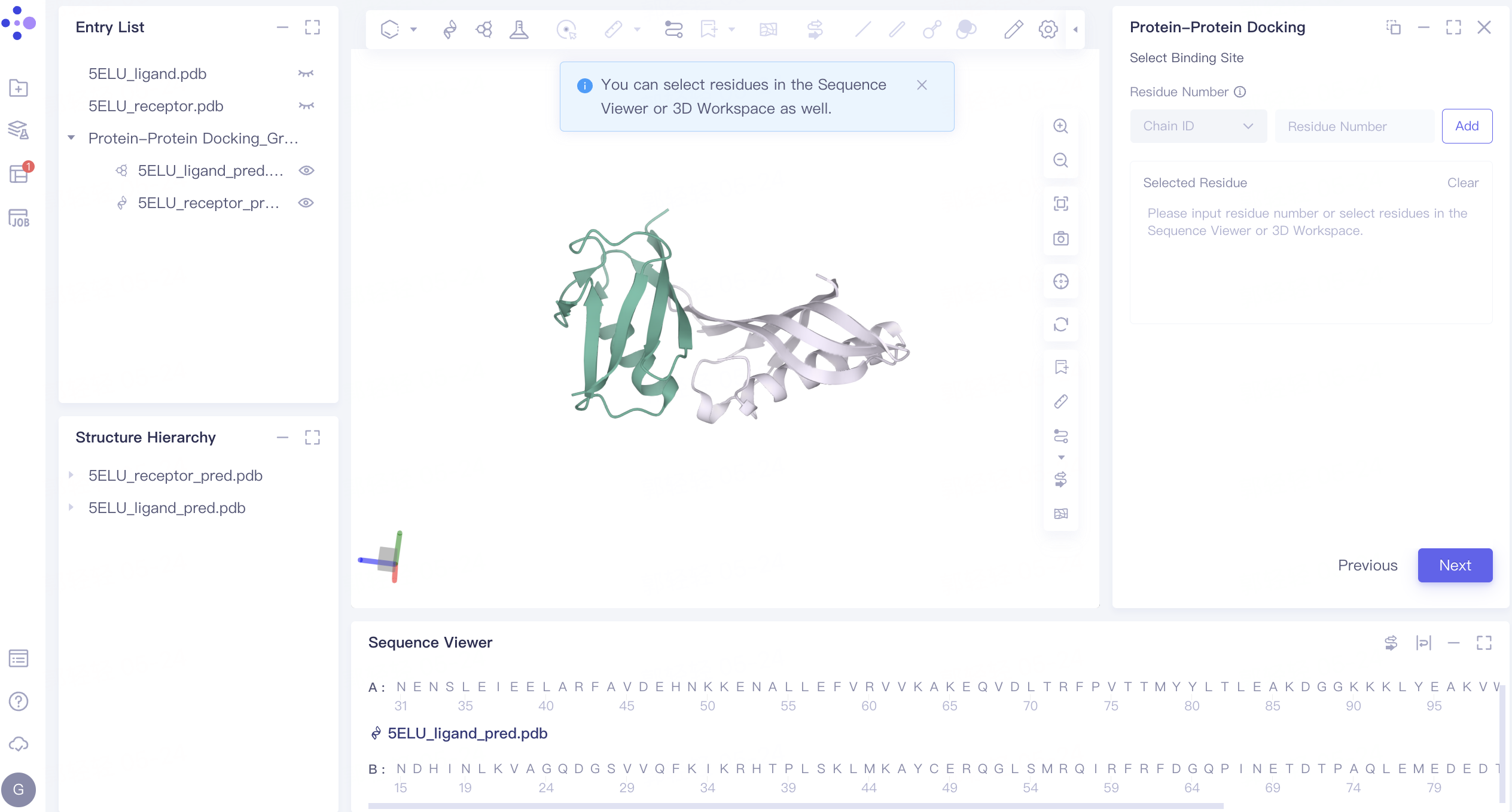
2.4 Binding site setting
- The residue of the binding site can be set for restriction according to the information of the relevant binding site. The binding site is not set here. Click 'Next' to enter the next step.
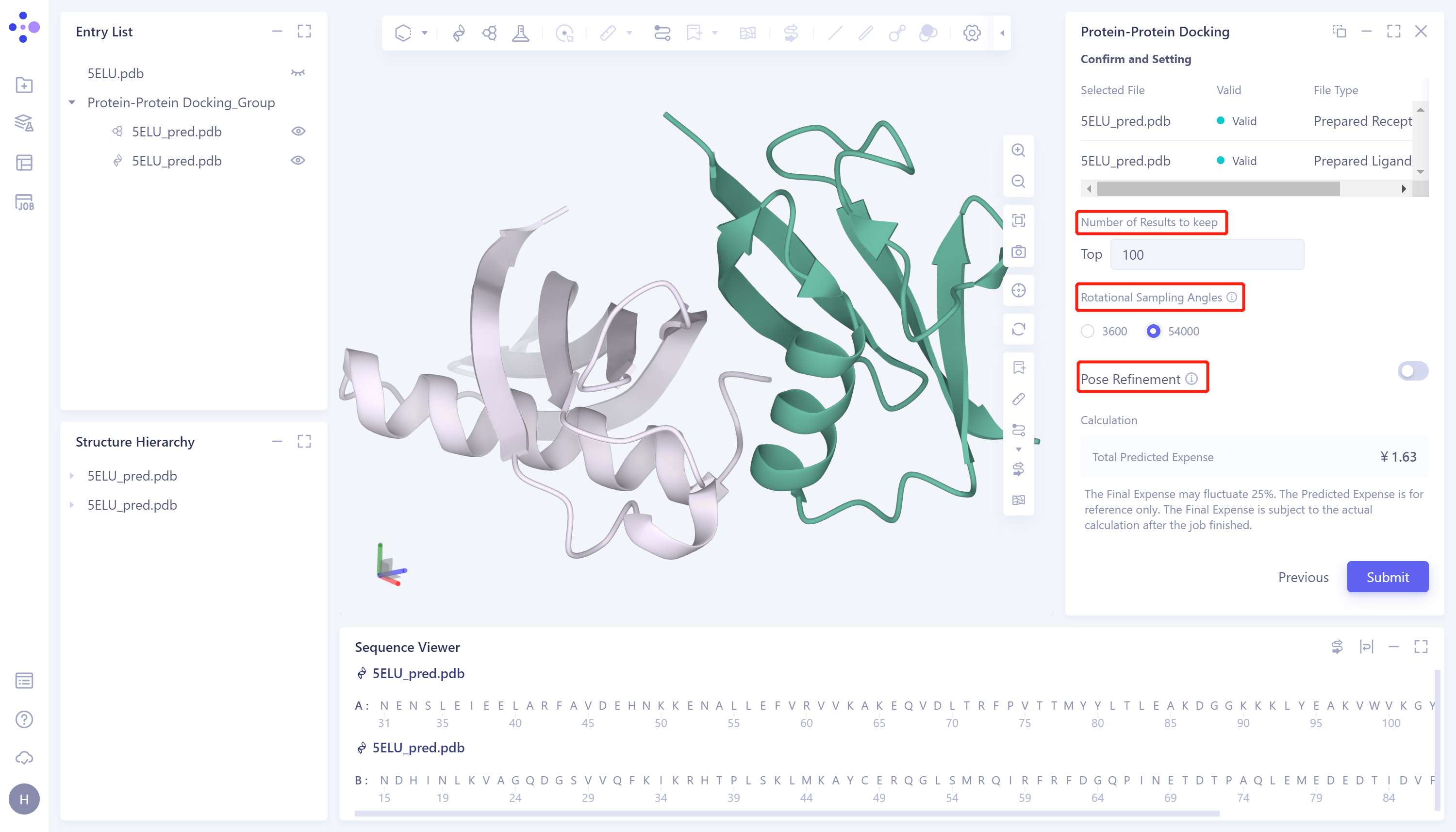
2.5 Parameter setting
- Confirm whether the receptor protein and ligand protein in the table are correct → Number of Results to keep: set the number of reserved results, which is set by the default value (100) → Rotational Sampling Angles: set the rotation sampling angle. Here, select 54000 → Pose Refinement: Optimize the conformation, and do not perform conformation optimization here → Total Predicted Expense, predict the cost consumed by the task → Name the job as 'PP docking' → click 'Submit' to submit the task.
2.6 Analysis of results
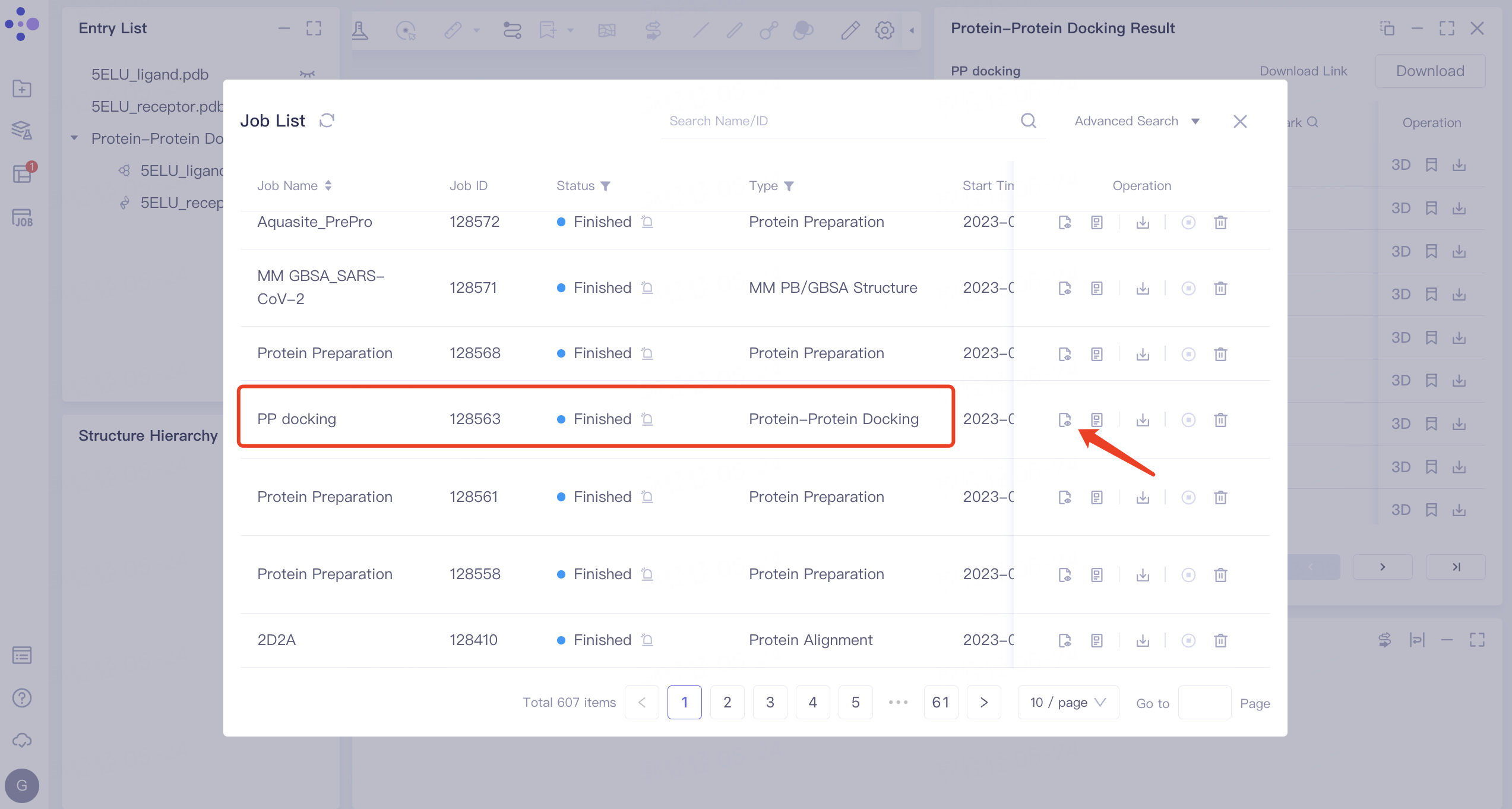
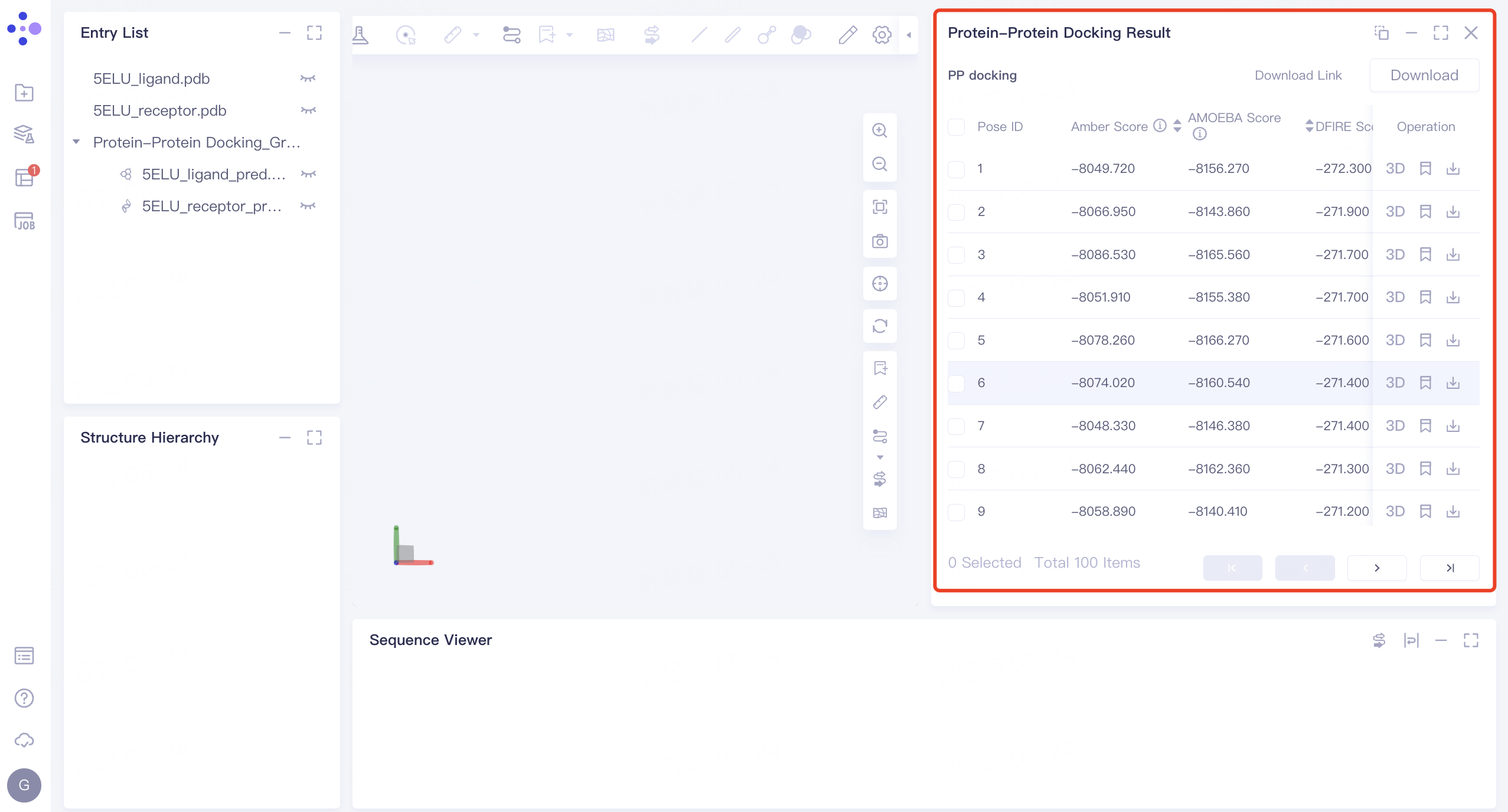
2.6.1 Inlet
- On the left side of the general menu bar, click 'Job' → find the task 'PP docking' → click 'Show' in the Operation column to display the results of the task → open the interface as shown in the right figure.
 |  |
2.6.2 Analysis of binding mode
Pose display
- Click the arrow next to Score in the 'Protein-Protein Docking Result' interface on the right to sort the Score (here, sort according to the 'DFIRE_Score'), and click '3D' in the Operation column. The binding mode of pose after ligand-receptor docking with the lowest DFIRE_Score is displayed in 3D Workspace.
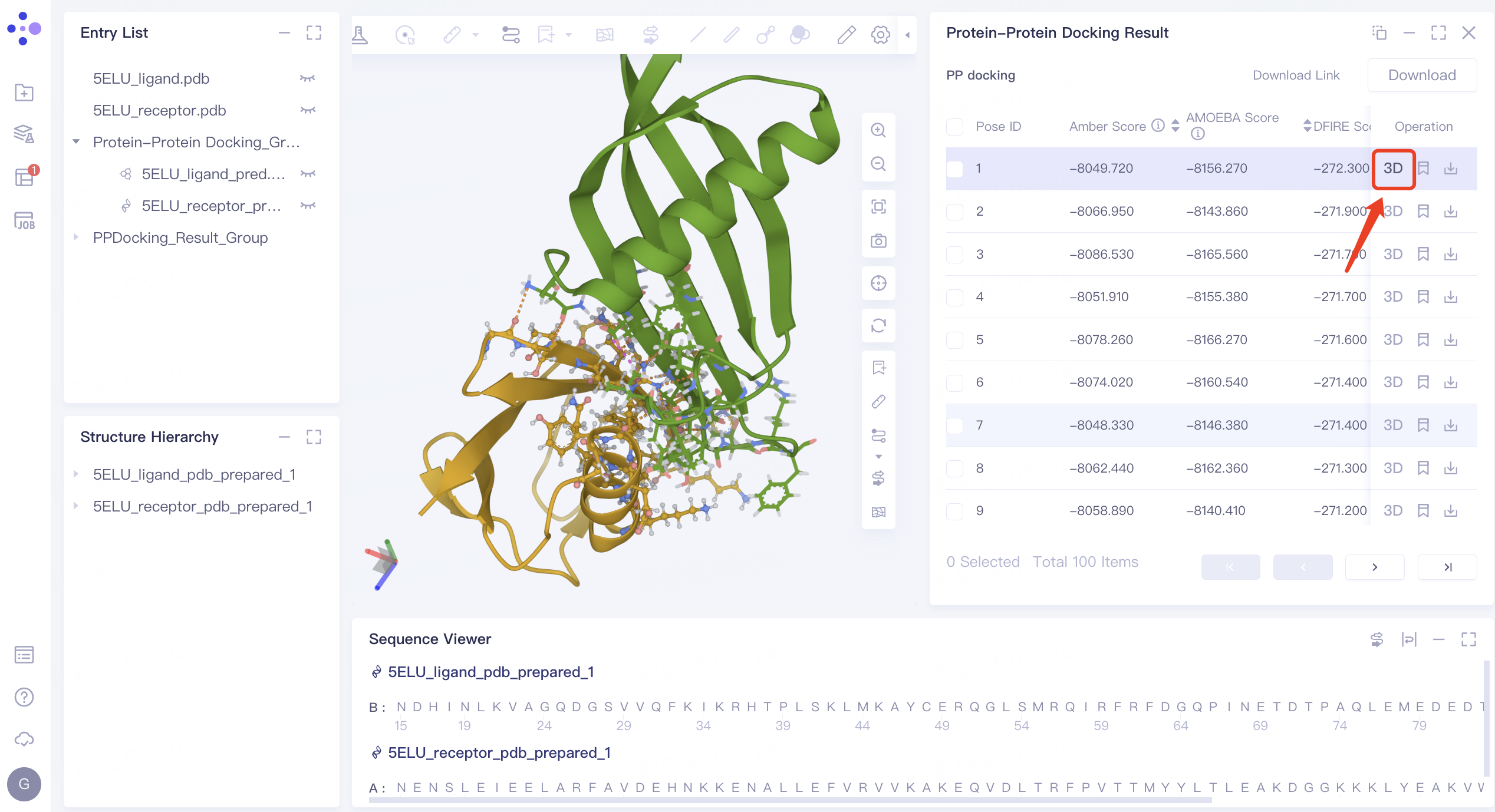
View non-bonding interactions
-
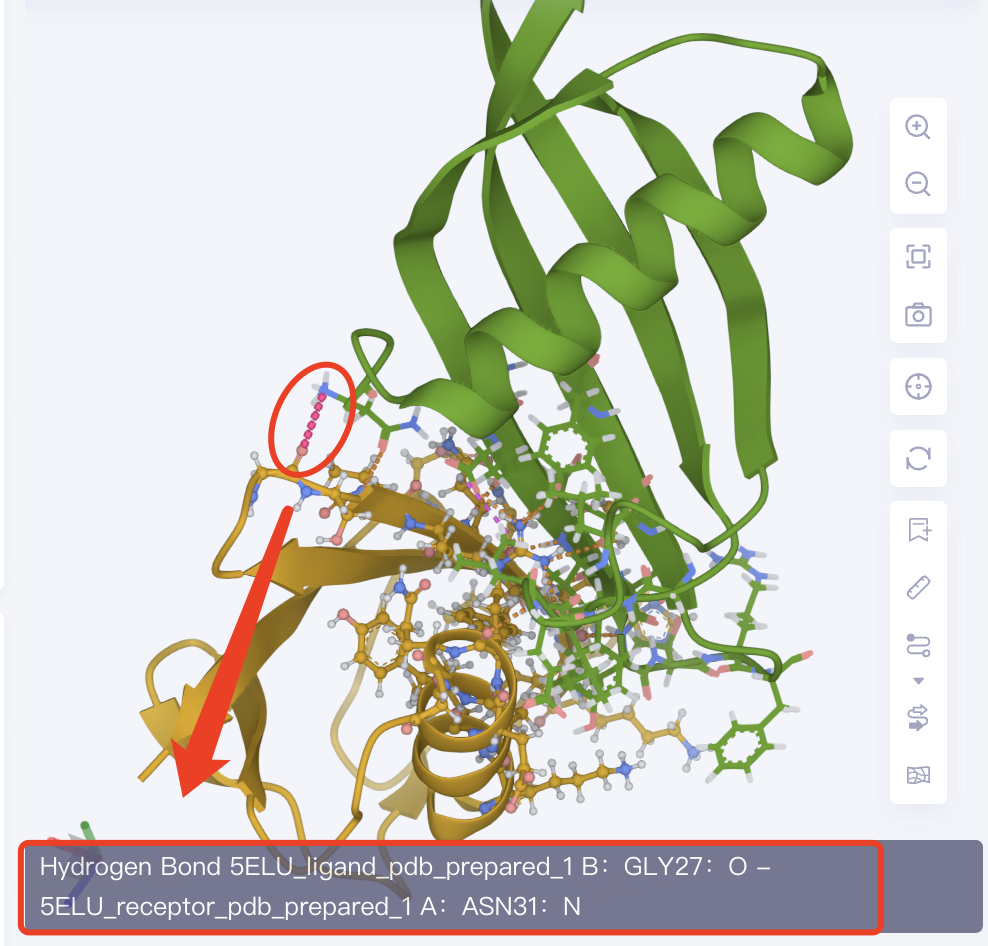
The amino acid residues of the ligand-receptor interaction interface are presented in the form of Line. The dotted line is the non-bonding interaction. Click the dotted line to show the specific description of the interaction in the lower left corner of the 3D Workspace.
-
Non-bond interactions are displayed by default, and the display of non-bond interactions can be controlled by Interaction in the toolbar on the right side of the 3D Workspce window. Click the arrow below Interaction to give the type of non-bond interaction and its color information.
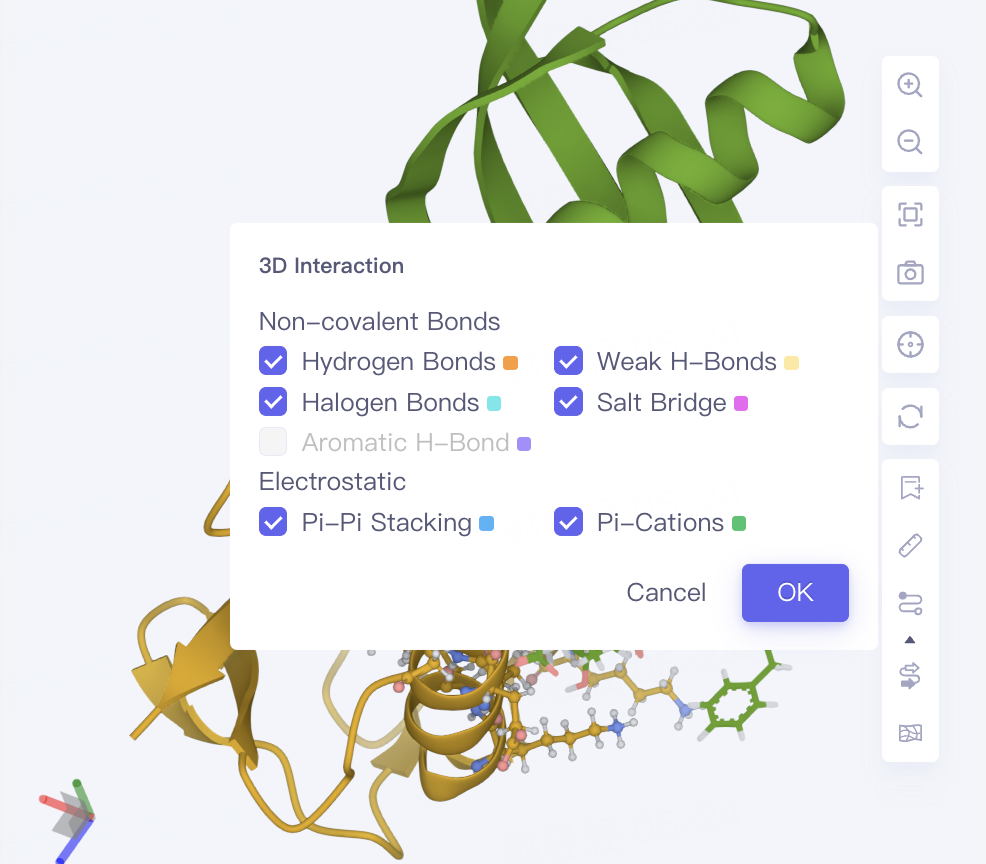
3. Evaluation of docking accuracy
3.1 Download Receptor and Ligand Protein Structure
- Download the structure of receptor protein and ligand protein with Pose ID 1: check the checkbox before pose 1 → click 'Download' in the upper right corner → check 'Merged Complex (.pdb)' in the pop-up 'Download' window, This file documents the structure of the pose 1 ligand protein and receptor protein complex → Click 'Submit'.
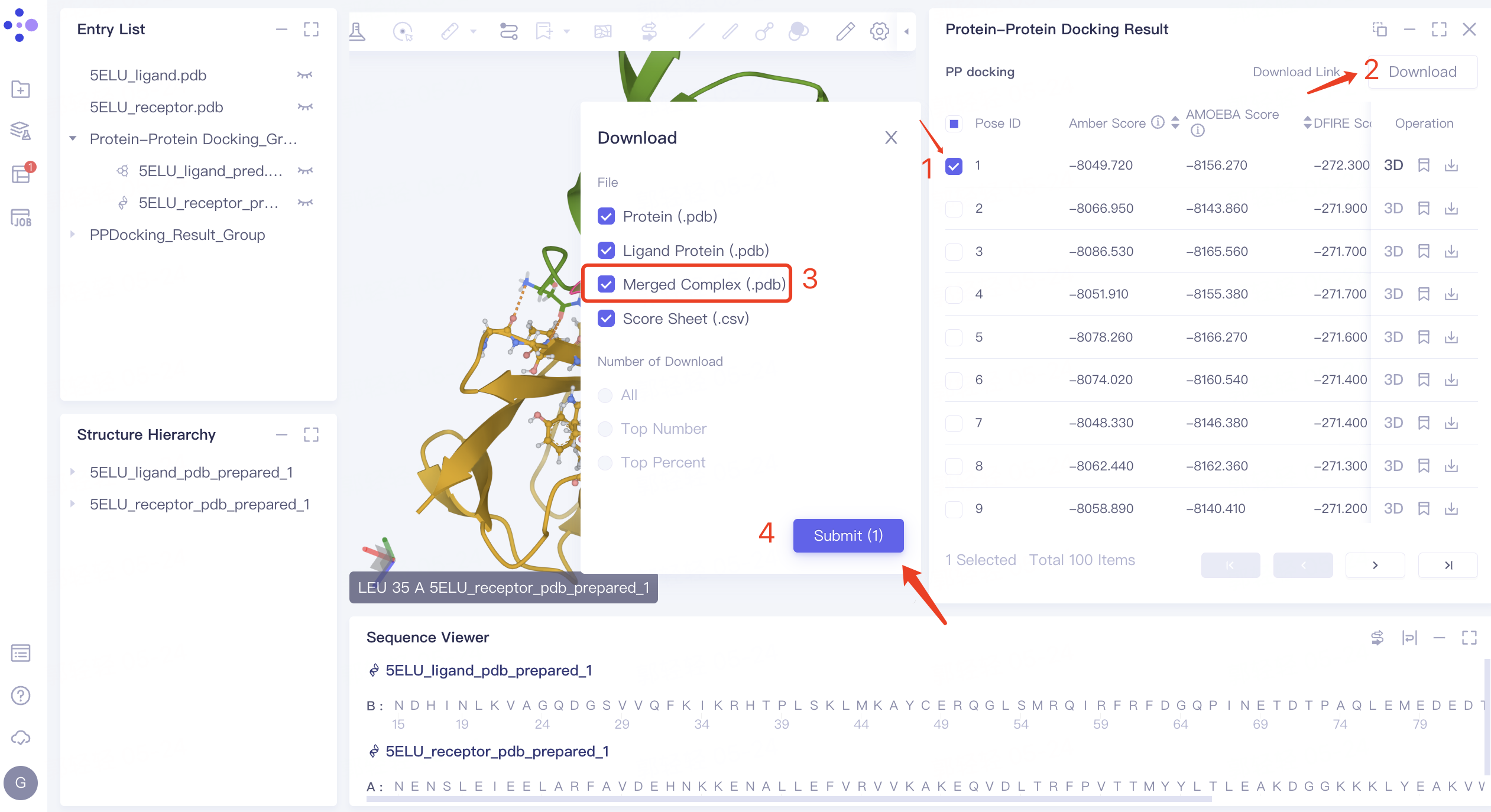
- The 'Download Link' flashes to show that the download link is ready. Click 'OK'.
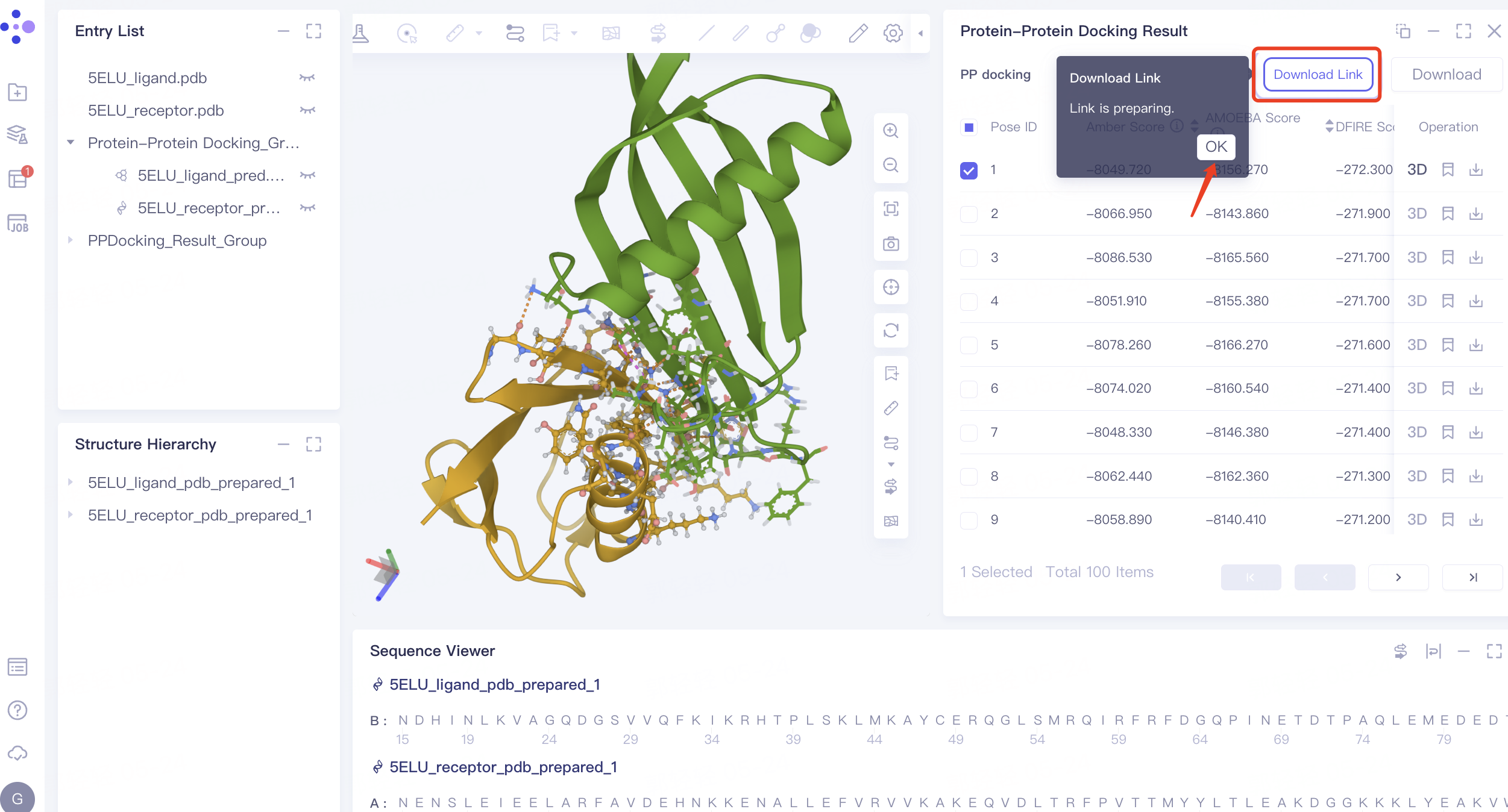
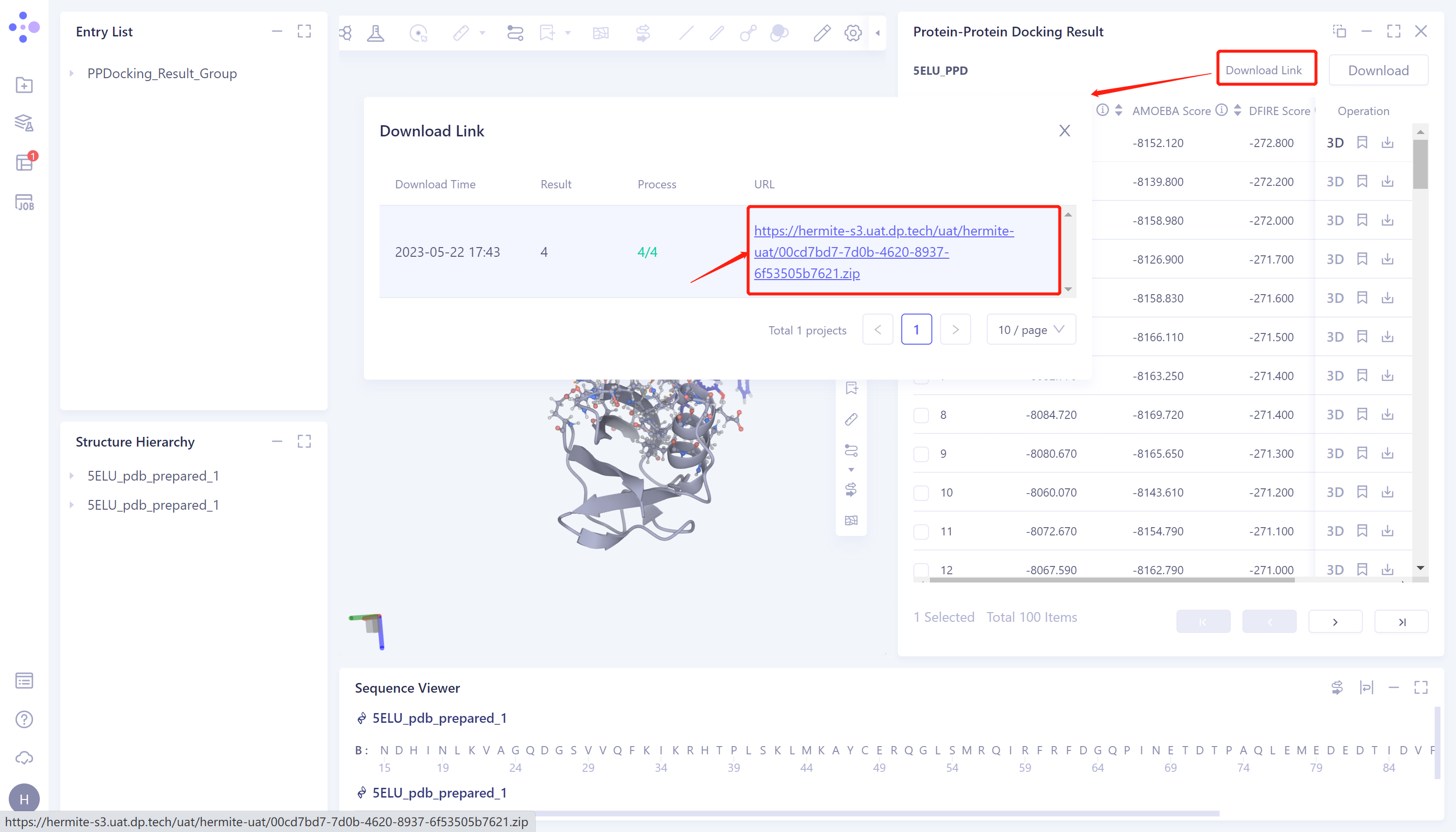
- Click 'Download Link' → The 'Download Link' window pops up → Click the link under the URL to download the file.
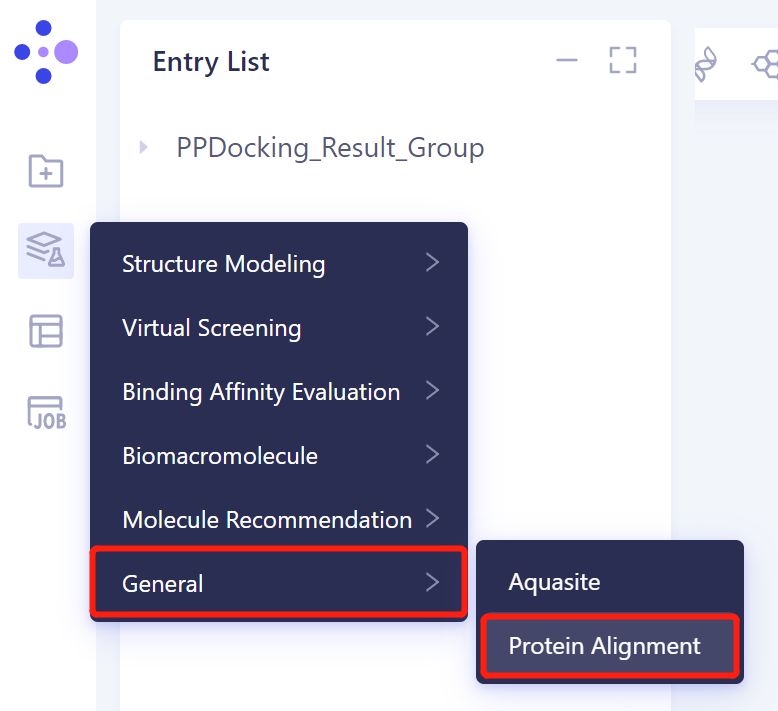
3.2 Protein Alignment Portal
- In the same project, click 'Function' → 'General' → 'Protein Alignment' in the left general menu bar.
3.3 Import the protein structure into the 3D Workspace window
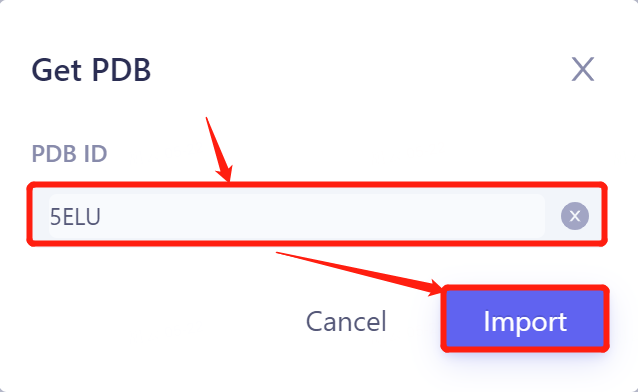
- In the general menu bar on the left, click 'File' → Get PDB → Input '5ELU' in the pop-up window to enter PDB ID: 5ELU , and click 'Import' to import the protein structure.
3.4 Introduction of reference protein structure
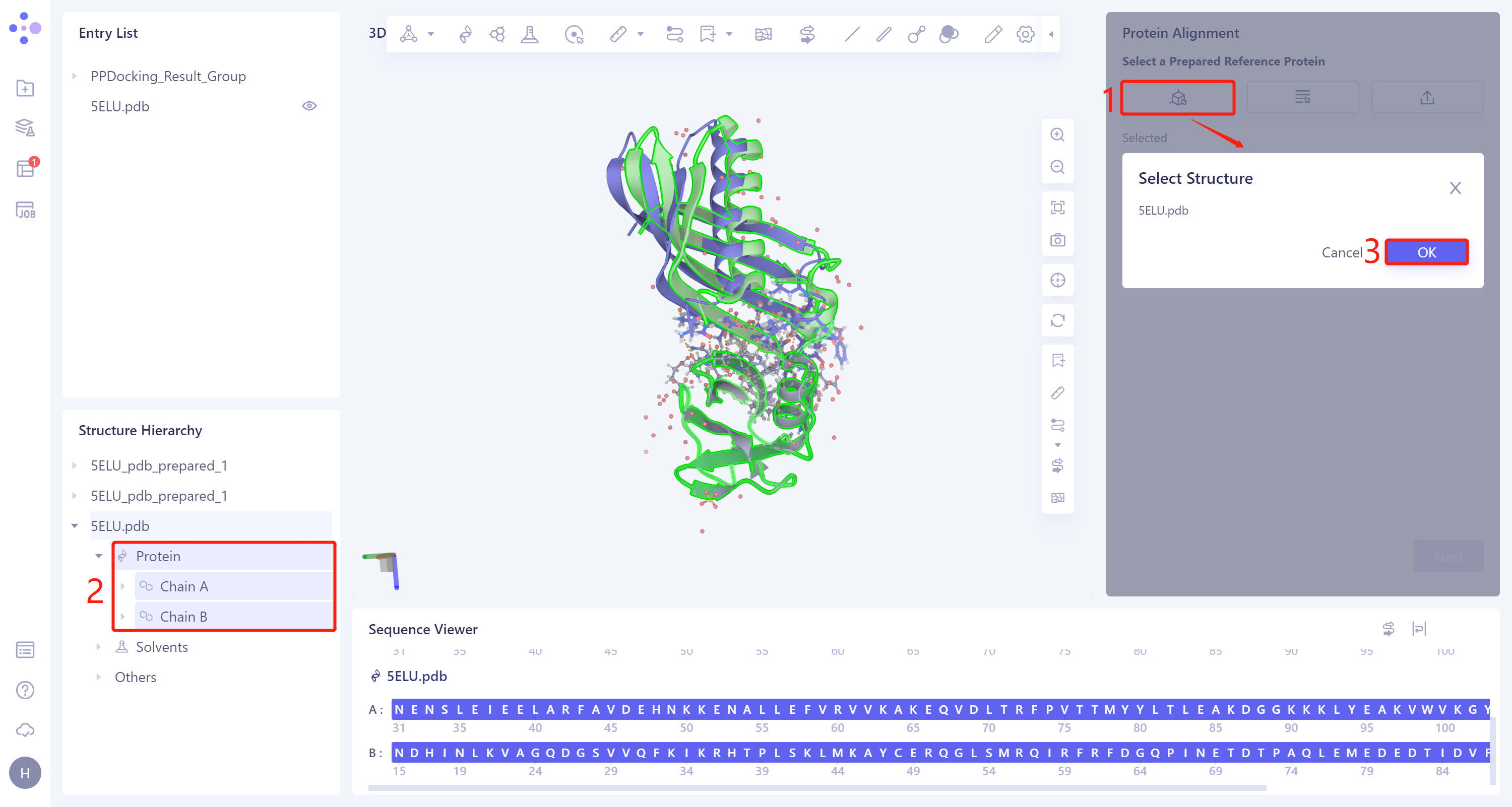
- The Protein Alignment operation window appears on the right side of the interface. First, import the reference protein structure: click 'Select Structure', The 'Select Structure' window pops up → Select the '5ELU' protein structure in the Structure Hierarchy → Click 'OK' in the 'Select Structure' window.
3.5. Import of protein structure to be compared
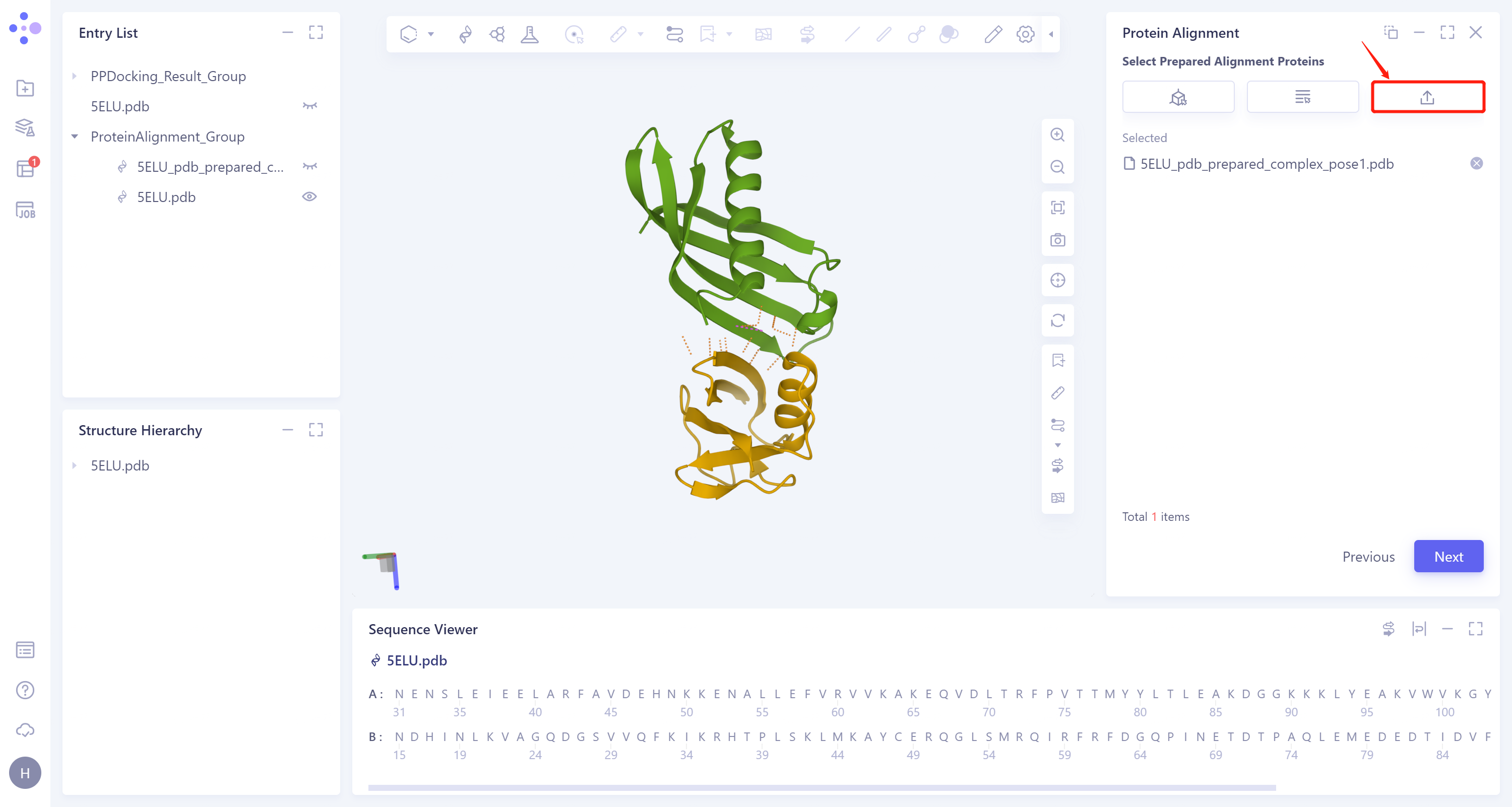
- Click 'Next' to enter the selection window of protein to be compared, import the protein structure to be compared: Click 'Select File', upload the local file (step 3.1 download the complex structure file in the file) '5ELU_pdb_ prepared_complex_pose1.pdb' → Click 'Next'.
3.6 Parameter setting
- Click 'Next' to enter the parameter setting page. 'Alignment Atoms' selects 'Cα' to perform alignment based on the α-carbon atoms between proteins. Since there is no special comparison requirement, the parameter setting of 'Select Binding Site' is skipped. The 'Job Name' field names the task as '5ELU_PA', and click 'Submit' to submit the task.
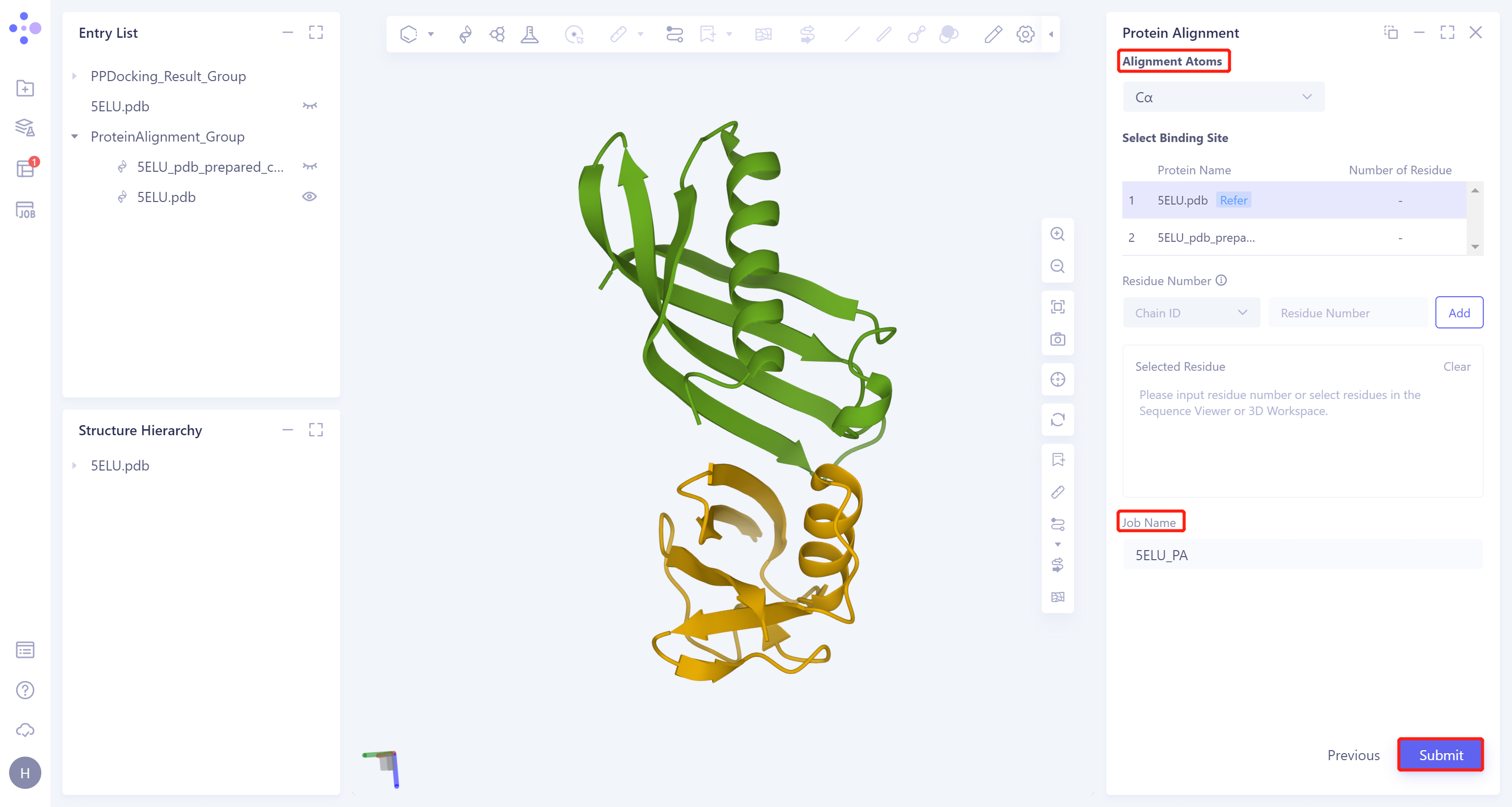
3.7 Analysis of results
3.7.1 Inlet
- General menu bar on the left: 'Job' → Find the task '5ELU_PA' in the pop-up 'Job List' window → Click 'Show' to display the task.
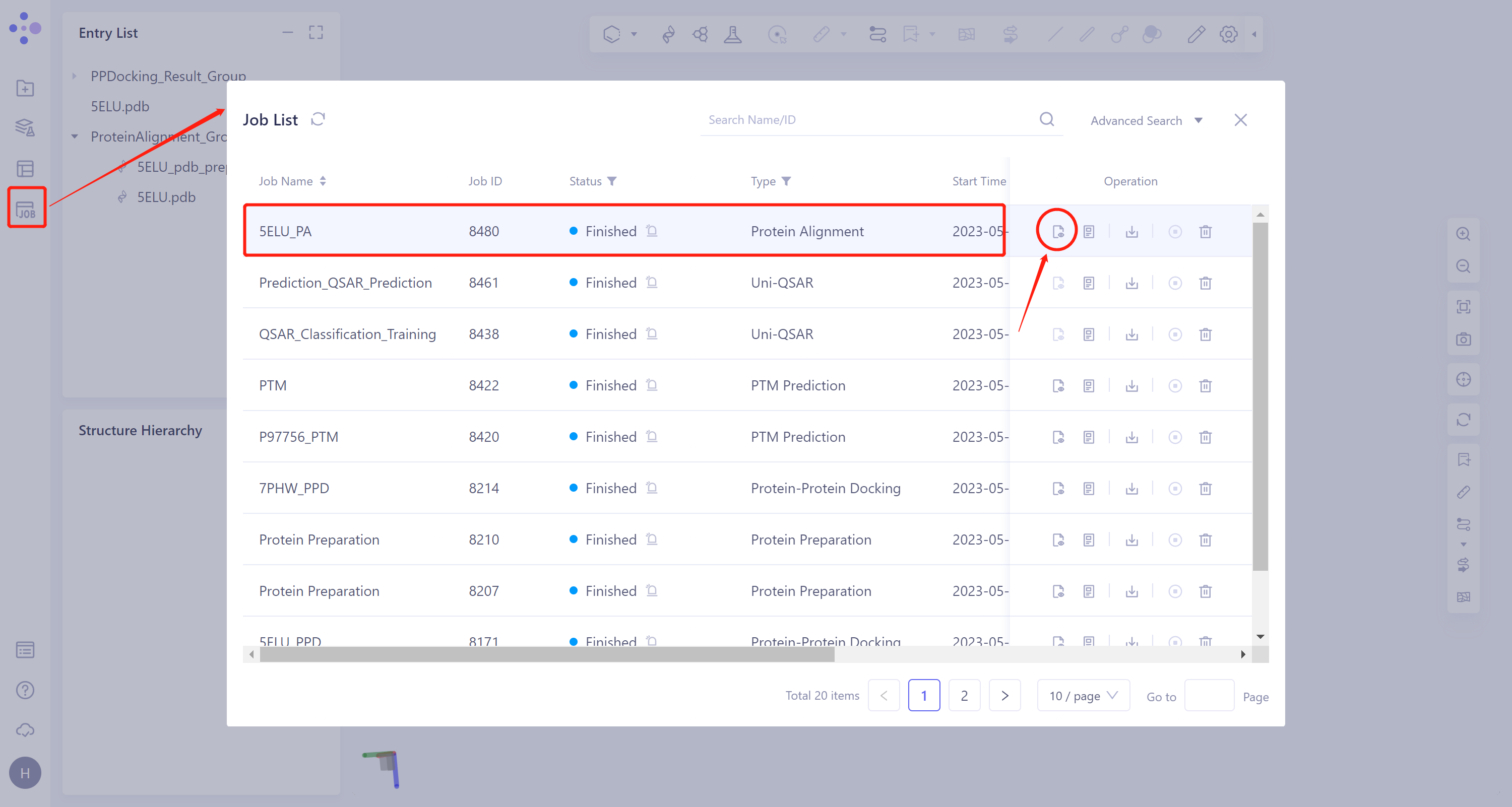
3.7.2 Presentation and explanation of results
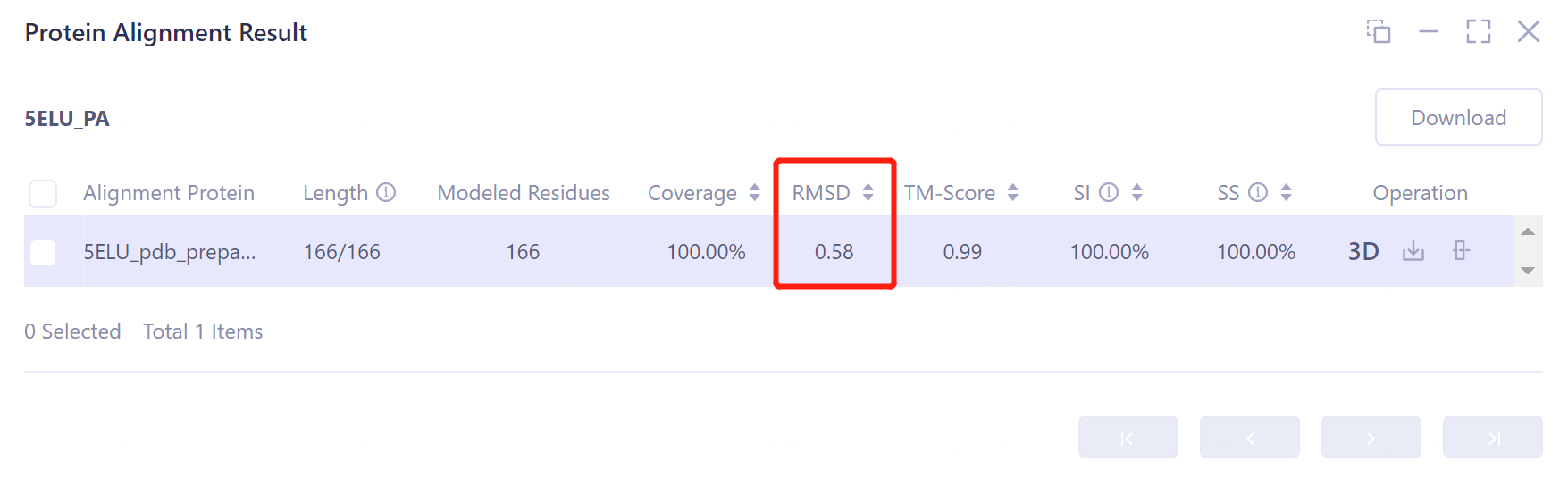
- The overlay image result of protein structure is displayed in the 3D Workspace window, and the 'Protein Alignment Result' window appears on the right side of the interface to display the protein alignment result.
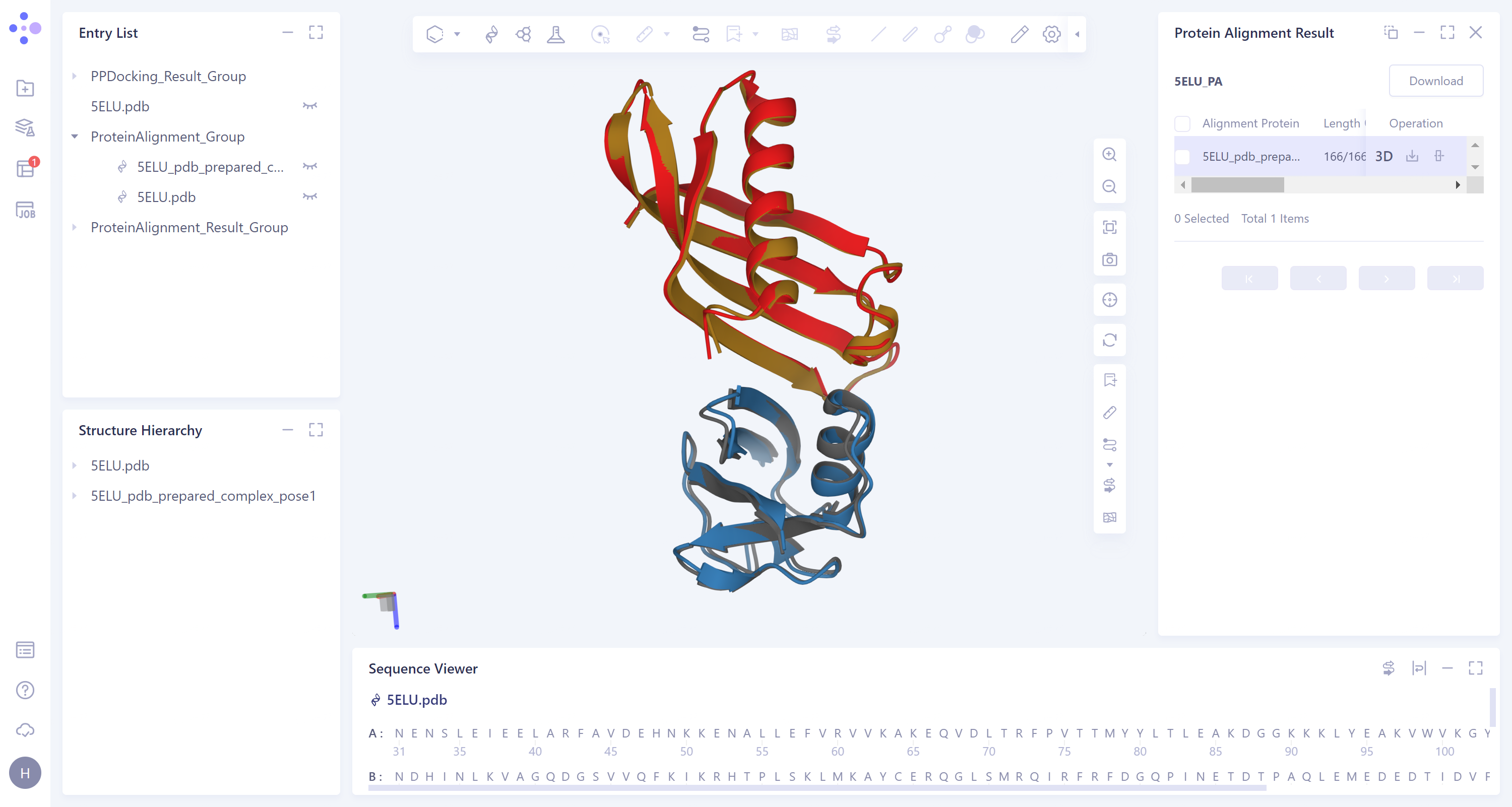
- After protein structure comparison, it was found that the RMSD value of the docked protein complex structure and the original protein complex structure was 0.58 Å, and the similarity was high, indicating that Protein-Protein Docking could reproduce the protein complex structure with high accuracy.