Prediction and Evaluation of Protein Multimer Structure Based on Uni-Fold
Introduction
For the first time, the Uni-Fold module in the Hermite ® platform successfully reproduces the full scale training of AlphaFold2, which can realize the structure prediction of protein monomers and protein polymers. In this tutorial, you will learn to use the Uni-Fold module to realize the structure prediction of protein multimers, and evaluate the accuracy of protein structure prediction by calling the Protein Alignment module in Hermite to compare the predicted structure with the real experimental structure.
7MLH is the X-ray crystal structure of the Fab complex of the house dust mite allergen Der P2 and human IgE monoclonal antibody (mAb) to a resolution of 2.10 �Å. This tutorial takes 7MLH as an example to predict its structure based on the sequence of the protein complex and evaluate the accuracy of the results.
The data required for this tutorial is as follows:
1. Protein Folding
1.1 Entrance
- The left general menu bar 'Function' → Structure Modeling → Protein Folding.
1.2 protein sequence introduction
- Import the protein sequence in the Protein Folding window that appears on the right (shown in red). Click 'Fasta File' shown in the red box to upload the sequence file of 7MLH protein (.fasta format). After uploading, the sequence information of the protein is automatically displayed in the sequence box corresponding to each chain.
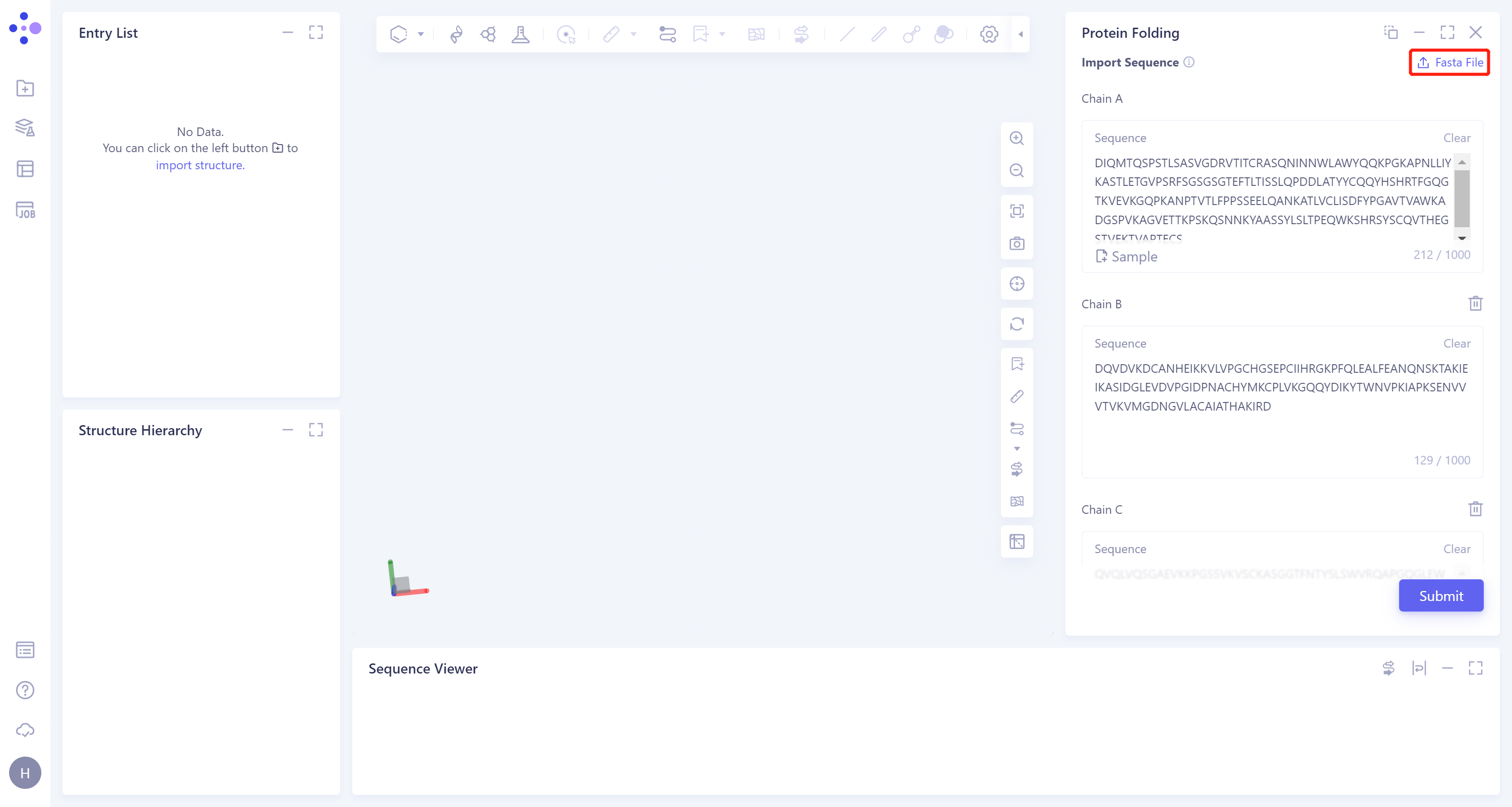
- After naming the task as 7MLH_Uni-Fold at the 'Job Name', click 'Submit' to submit the task.
1.3 Results View
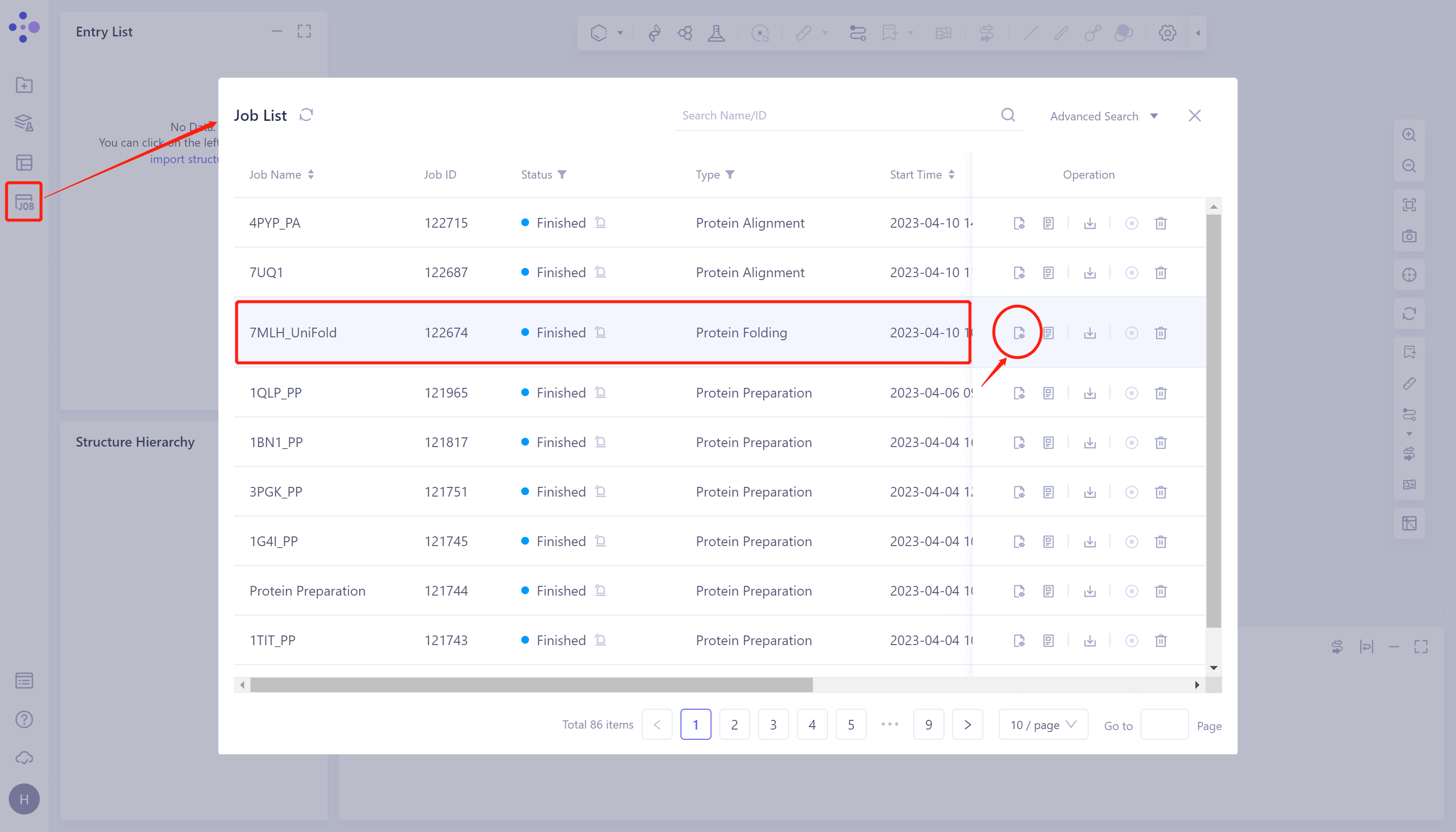
- Click 'Job' in the left general menu bar → find the 7MLH_Uni-Fold task in the pop-up 'Job List' window → click 'Show'.
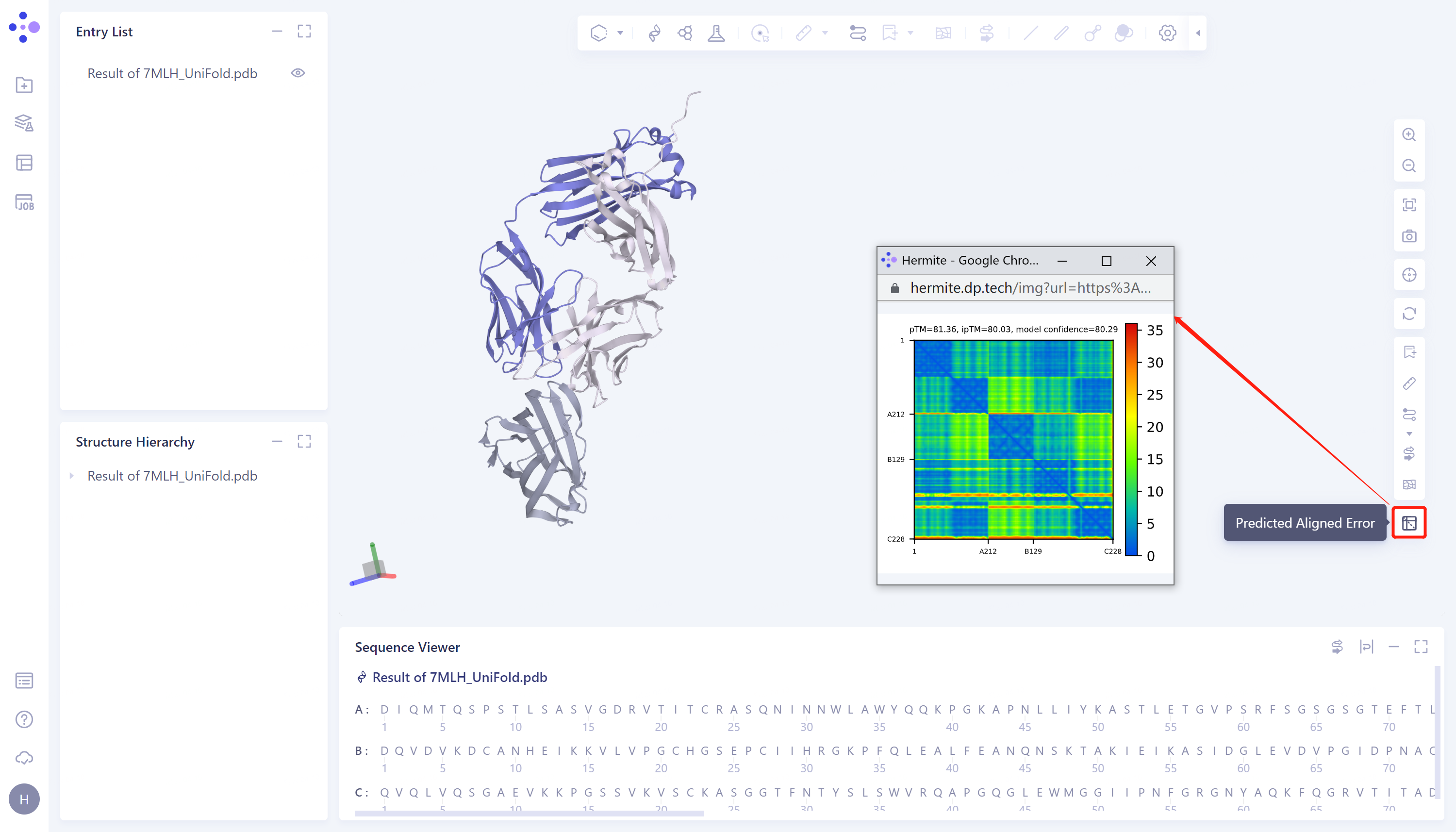
- The protein prediction results were displayed in the 3D Workspace, and the predicted aligned error on the right side was the evaluation of the prediction results, with a confidence level of 80.29.
2. Protein Alignment
2.1 Import protein structures into the 3D Workspace window
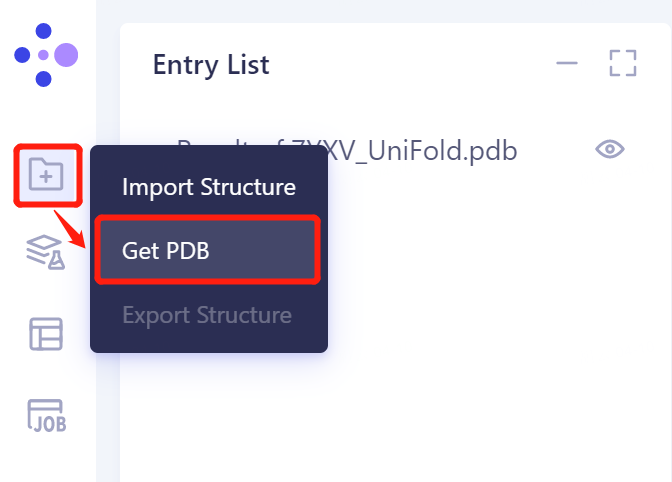
- The left general menu bar 'File' → Get PDB.
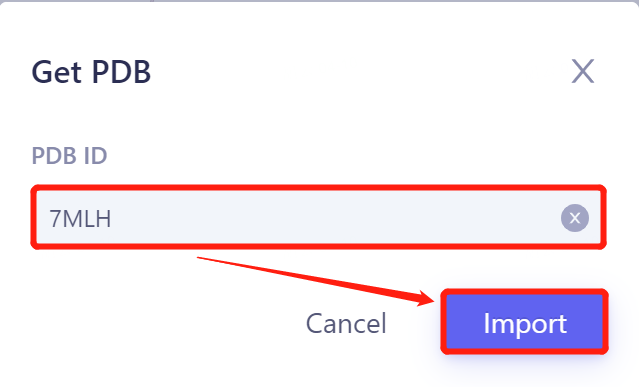
- Input '7MLH' in the pop-up window and click 'Import' to import the protein structure.
2.2 Invoke Protein Alignment module
2.2.1 Inlet
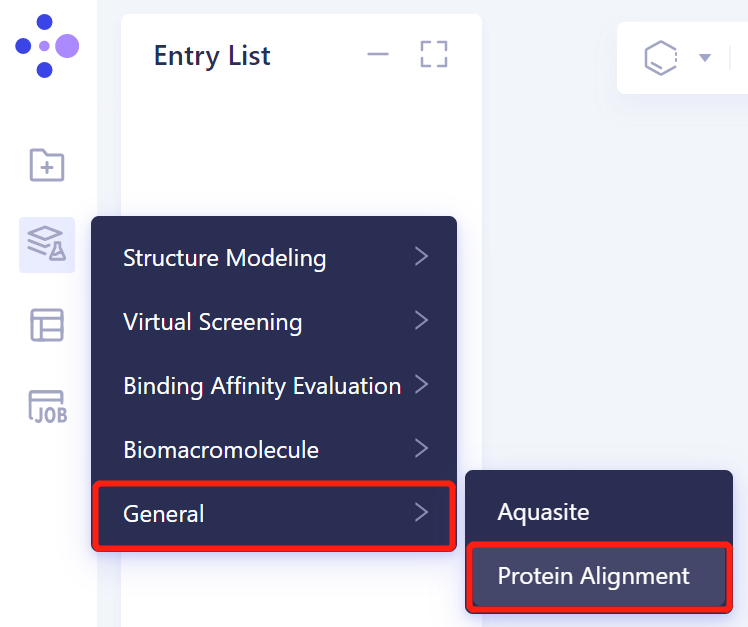
- In the same Project, click 'Function' → General → Protein Alignment on the left general menu bar.
2.2.2 Introduction of reference protein structures
- The 'Protein Alignment' interface appears on the right side of the interface. First, import the reference protein structure: click 'Select Structure', The 'Select Structure' interface pops up → Select the protein structure of '7MLH' in the Structure Hierarchy → Click 'OK' in the 'Select Structure' window.
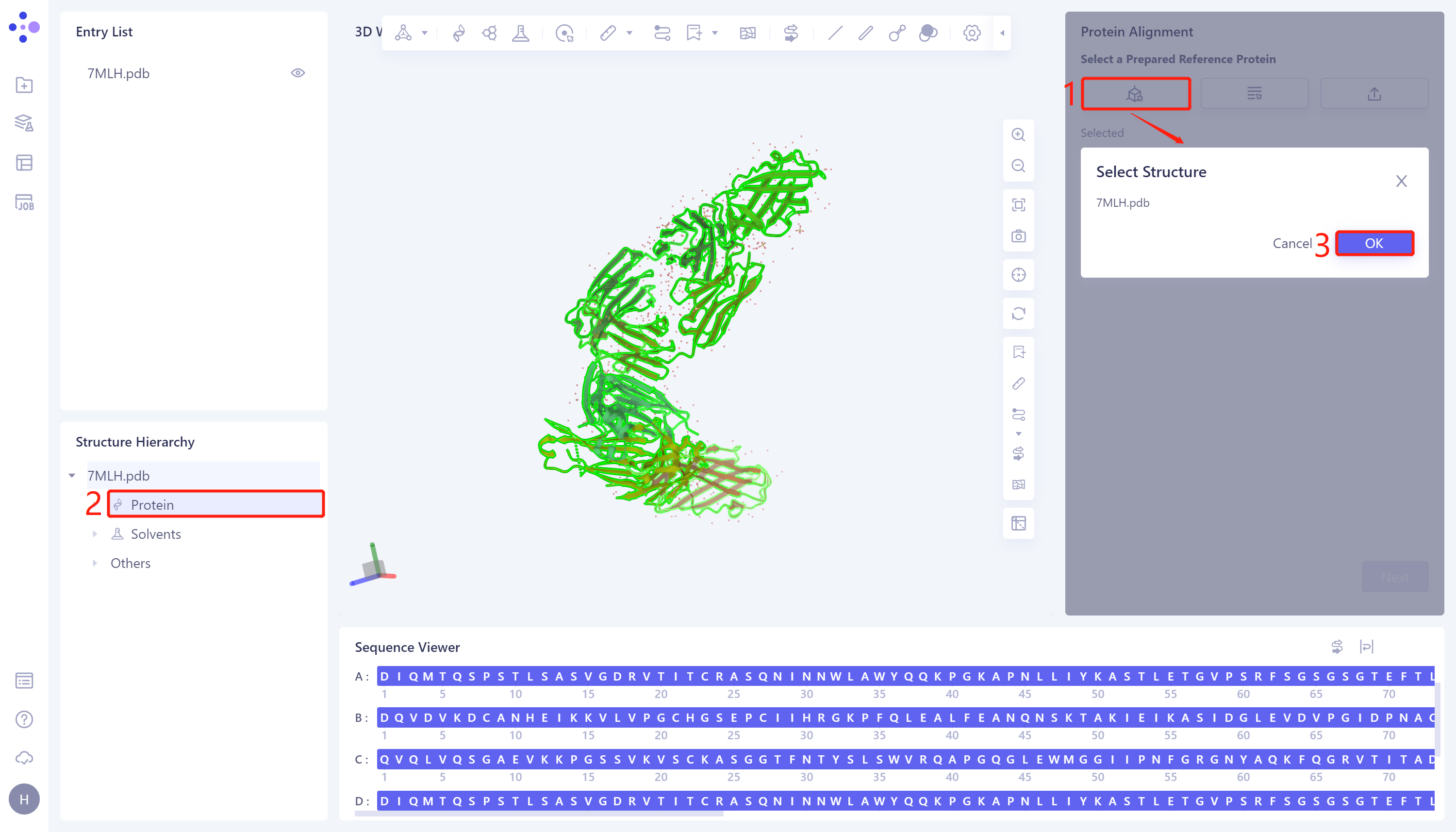
2.2.3 introduction of protein structure to be compare
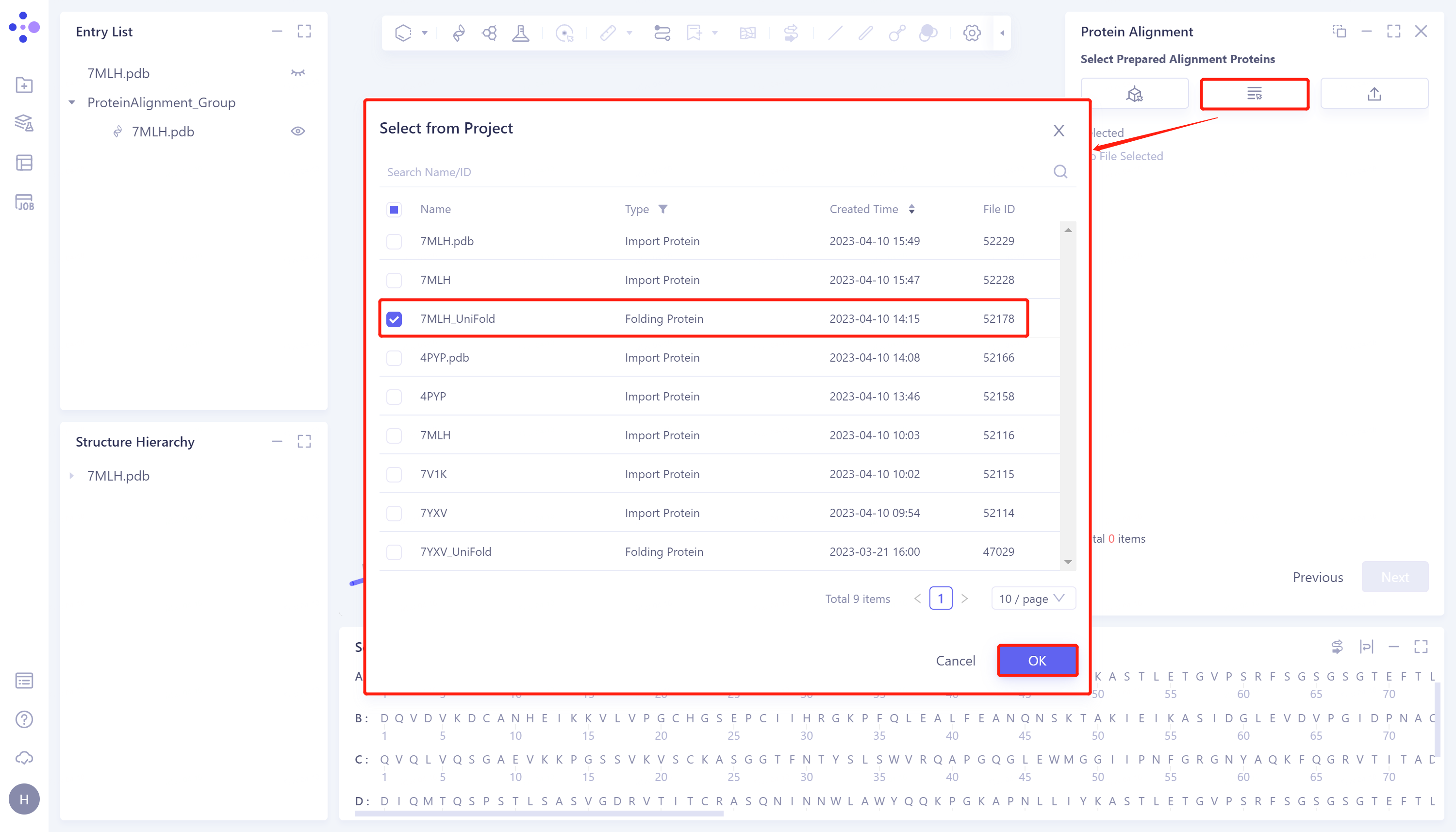
- Click 'Next' to enter the selection window of protein to be compared and import the protein structure to be compared: click 'Select from Project', The 'Select from Project' window pops up → Select '7MLH_Uni-Fold' in the 'Select from Project' window → Click 'OK'.
2.2.4 Parameter setting
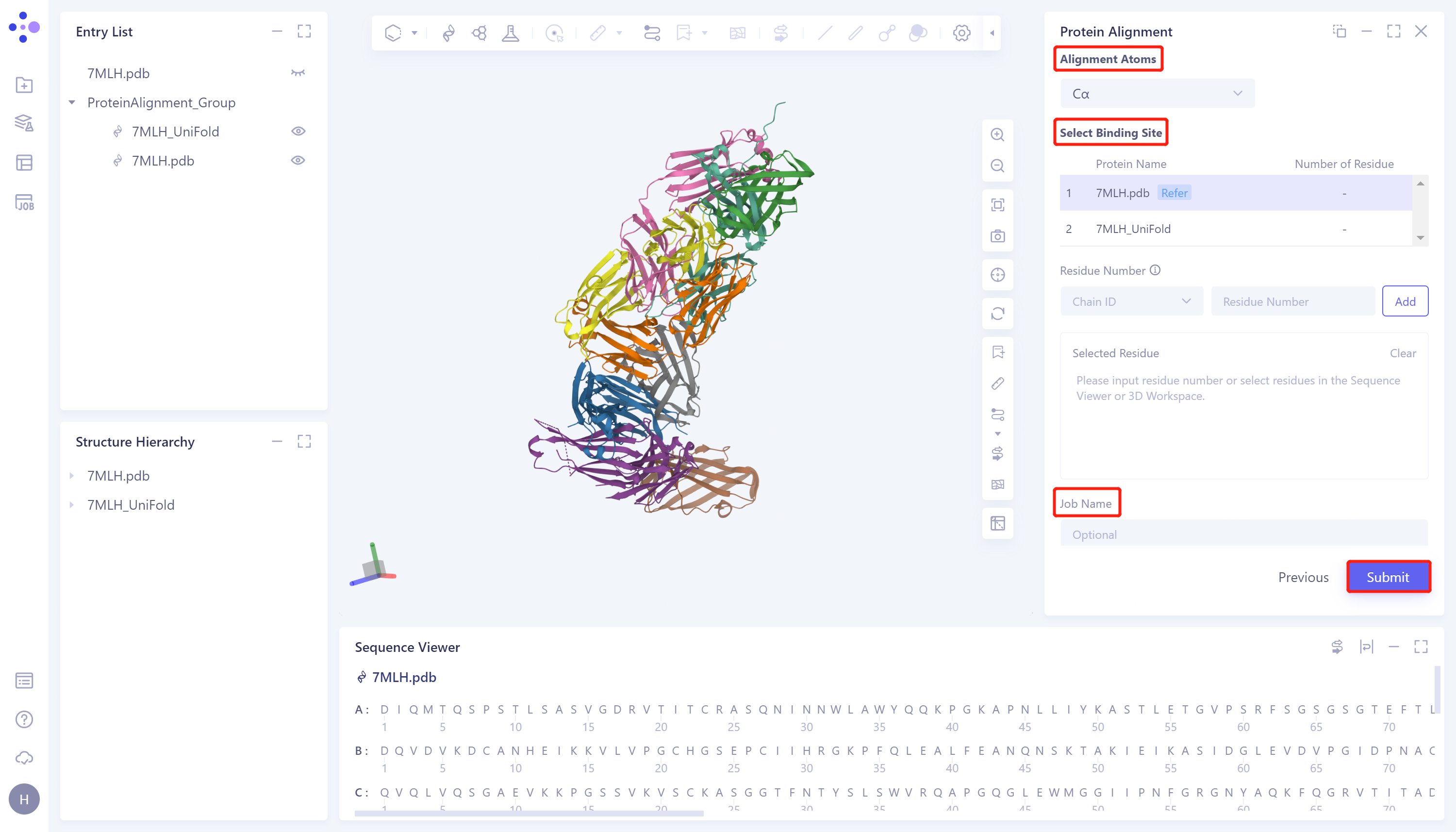
- Click 'Next' to enter the parameter setting page. 'Alignment Atoms' selects 'Cα' to perform alignment based on the α-carbon atoms between proteins. Since there is no special comparison requirement, the parameter setting of 'Select Binding Site' is skipped. Name the task as '7MLH_PA' at the 'Job Name', and click 'Submit' to submit the task.
2.3 Analysis of results
2.3.1 Inlet
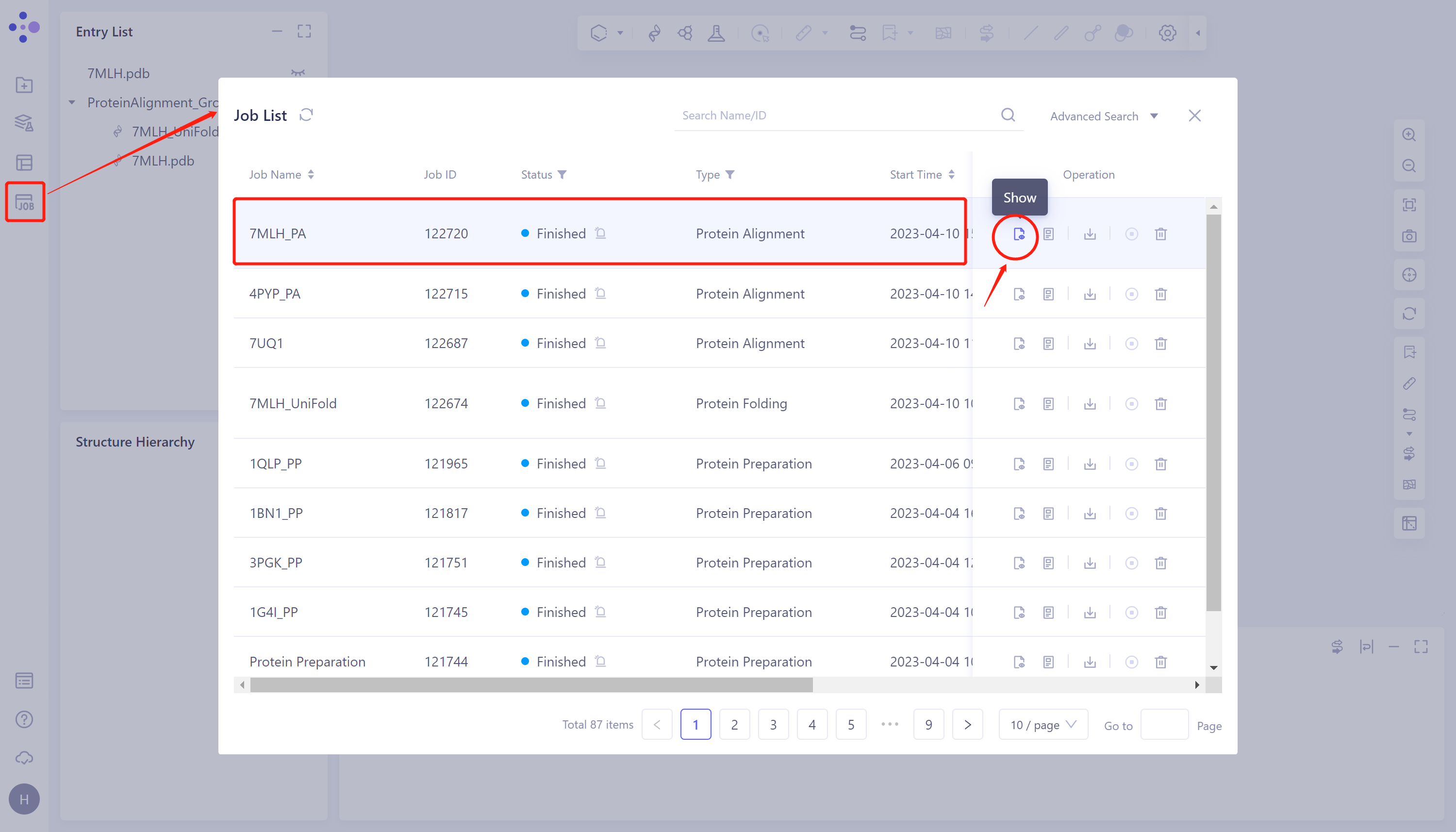
- General menu bar on the left: Job → Find task '7MLH_PA' in the pop-up 'Job List' window → Click 'Show' to display the task.
2.3.2 Presentation and explanation of results
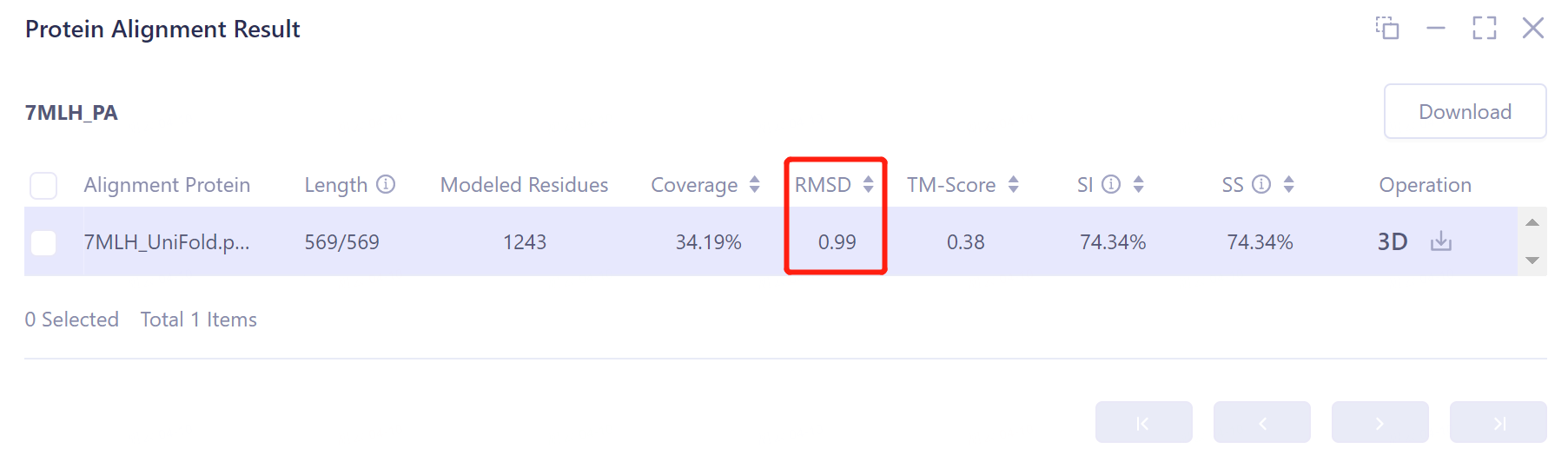
- The overlay image result of protein structure is displayed in the 3D Workspace window, and the 'Protein Alignment Result' window appears on the right side of the interface to display the protein alignment result.
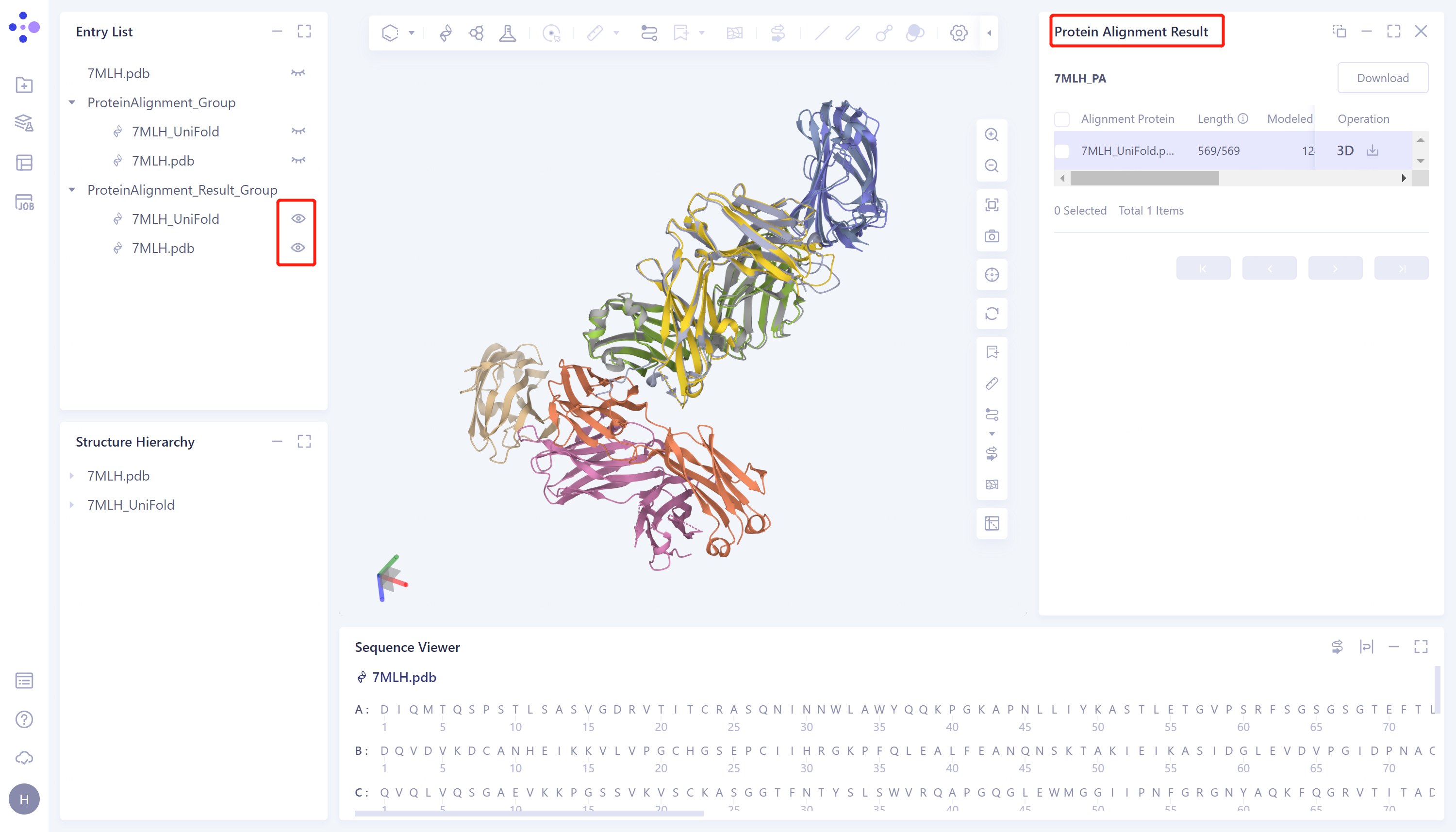
- Through protein structure comparison, it was found that the RMSD value of Uni-Fold predicted multimer structure and the original protein structure was 0.99 Å, and the similarity was high, indicating that Uni-Fold could well predict the structure of protein multimer 7MLH.