Virtual Screening Workflow
简介
虚拟筛选在药物设计中具有重要作用,它可以提高药物研发效率、指导合成和优化、提高命中率、发现新的药物靶点以及进行药物再定位等。
Hermite平台的Virtual Screening Workflw模块提供了高通量虚拟筛选功能,集成了依据配体化学性质的过滤工具、四种限制性对接(H-Bond、Maximum Common Structure、Substructure、Shape)、基于对比学习算法粗筛配体的Uni-Clip、速度精度大大提升的分子对接工具Uni-Dock[1]、优化对接pose的Uni-Mol预训练模型以及计算结合自由能的MM PB/GBSA,您可全面快速地完成虚拟筛选相关工作。并且,依托于简单易上手的操作界面,您可以快速掌握计算机辅助药物设计中的虚拟筛��选方法。此外,Hermite平台内置海量虚拟化合物库(2200w),您可以快速完成大化学空间的虚拟筛选任务。
σ2受体在增殖细胞和许多肿瘤中过表达,标记的σ2配体已被提出作为癌症诊断和治疗的工具。σ2受体也被认为是治疗中枢神经系统疾病的靶点。7M94为σ2受体与Roluperidone(一种σ2受体拮抗剂)的共晶配体[2],本教程基于该受体对数据库中的分子进行虚拟筛选。
本教程所需文件如下:
1. Virtual Screening Workflow
1.1 功能入口
左侧通用菜单栏Function → Virtual Screening → Virtual Screening Workflow。
弹出“MM PB/GBSA”窗口,点击“Yes”。
1.2 Select a Prepared Protein
点击“Select File”,上传本地文件“7M94_prep.pdb”;
上传的蛋白自动进入力场检查,待Valid处由“Processing”变成“Valid”后,点击“Next”。
注:力场检查不通过时,可通过右上角“Protein Preparation”进行蛋白准备。更多蛋白选择方式的操作说明,详见《蛋白及配体选择 》。
1.3 配体准备
非此处Database来源的配体,自动进入配体准备流程,详见《Ligand Preparation》。
1.3.1 Select Prepared Ligands From
点击“Select from Database”,弹出Select Ligands窗口 → 选择数据集“Specs”,点击“OK”。
点击“Next”。
注:在进行虚筛前,配体需要进行配体准备,包括生成同分异构体、加氢、质子化、能量最小化等。Hermite中的数据库中的配体已经过配体准备处理,因此选择“Database”中的配体数据集不进入配体准备的工作流。更多配体选择方式的操作说明,详见《蛋白及配体选择 》。
1.3.2 Filters Setting
根据化学性质的取值范围过滤配体,此处不设置。
| 化学性质 | 含义 |
| Molecular Weight | 分子质量 |
| Lipid-Water Partition Coefficient | 脂水分配系数 |
| Number of Hydrogen Bond Donors | 氢键供体数目 |
| Number of Hydrogen Bond Acceptors | 氢键受体数目 |
| Number of Rotatable Bonds | 可旋转键数目 |
1.4 Pocket Setting
点击“Select File��”按钮,上传本地盒子文件“pocket.txt”。
注:其他盒子生成方式见下表,盒子体积不得小于1000ų。更多口袋设置方式的操作说明,详见《Pocket 》。
1.5 Constrained Docking
在本教程中,不需要基于条件限制进行对接,因此此处点击“No, skip this step”,跳过该步骤。
注:若您需要进行基于条件的限制对接,可参考下表:
| Constrained Docking | 操作方式 | 界面展示 | 注 |
| H-Bond | 1) 选择“H-Bond”,Ligand Showed in 3D Workspace下拉选择参考配体,点击“Confirm”; 2)在3D Workspace窗口中选择与配体具有氢键相互作用的受体中的原子,选中的原子自动解析至右侧氢键受体和供体列表中 | 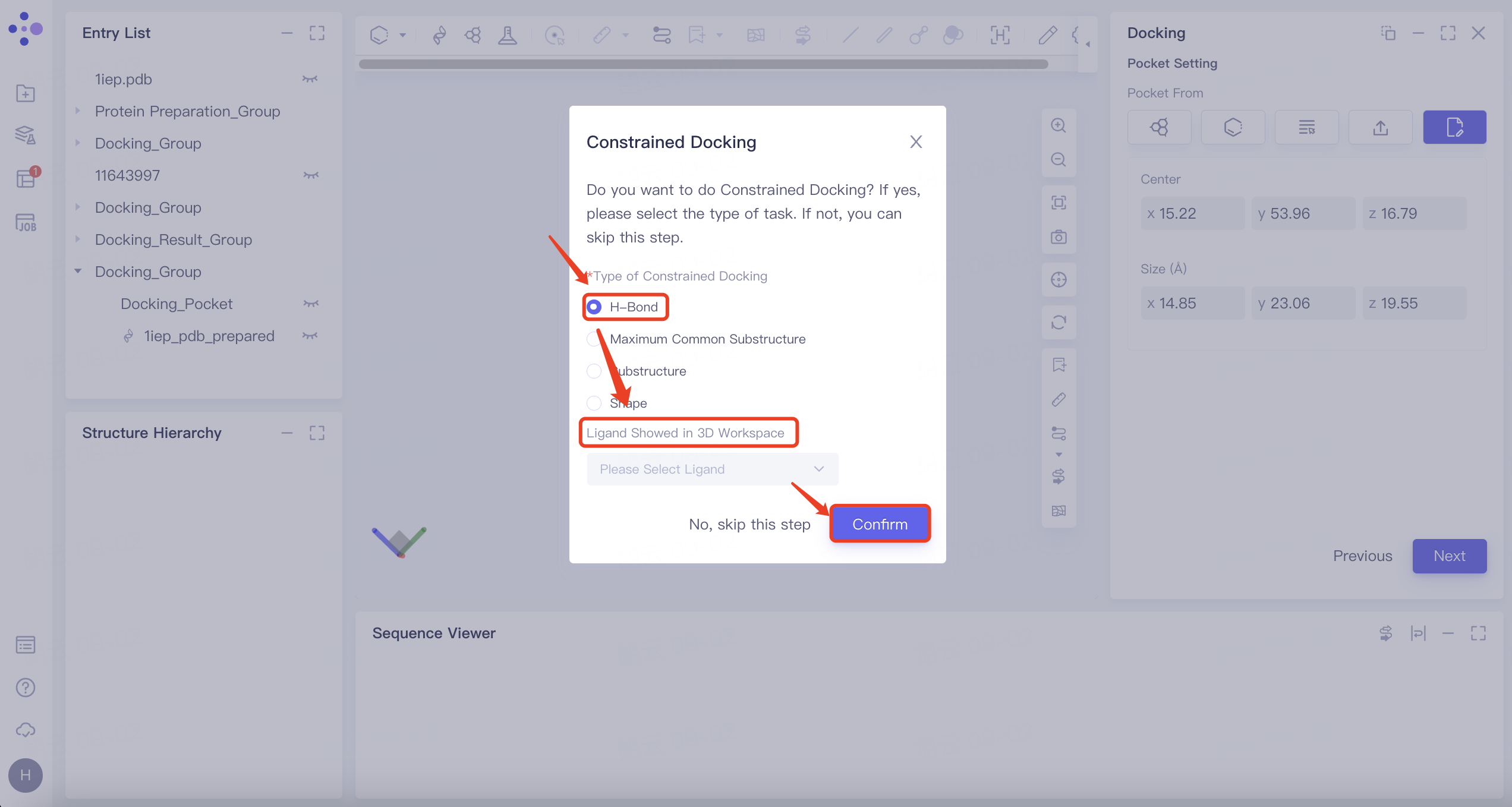 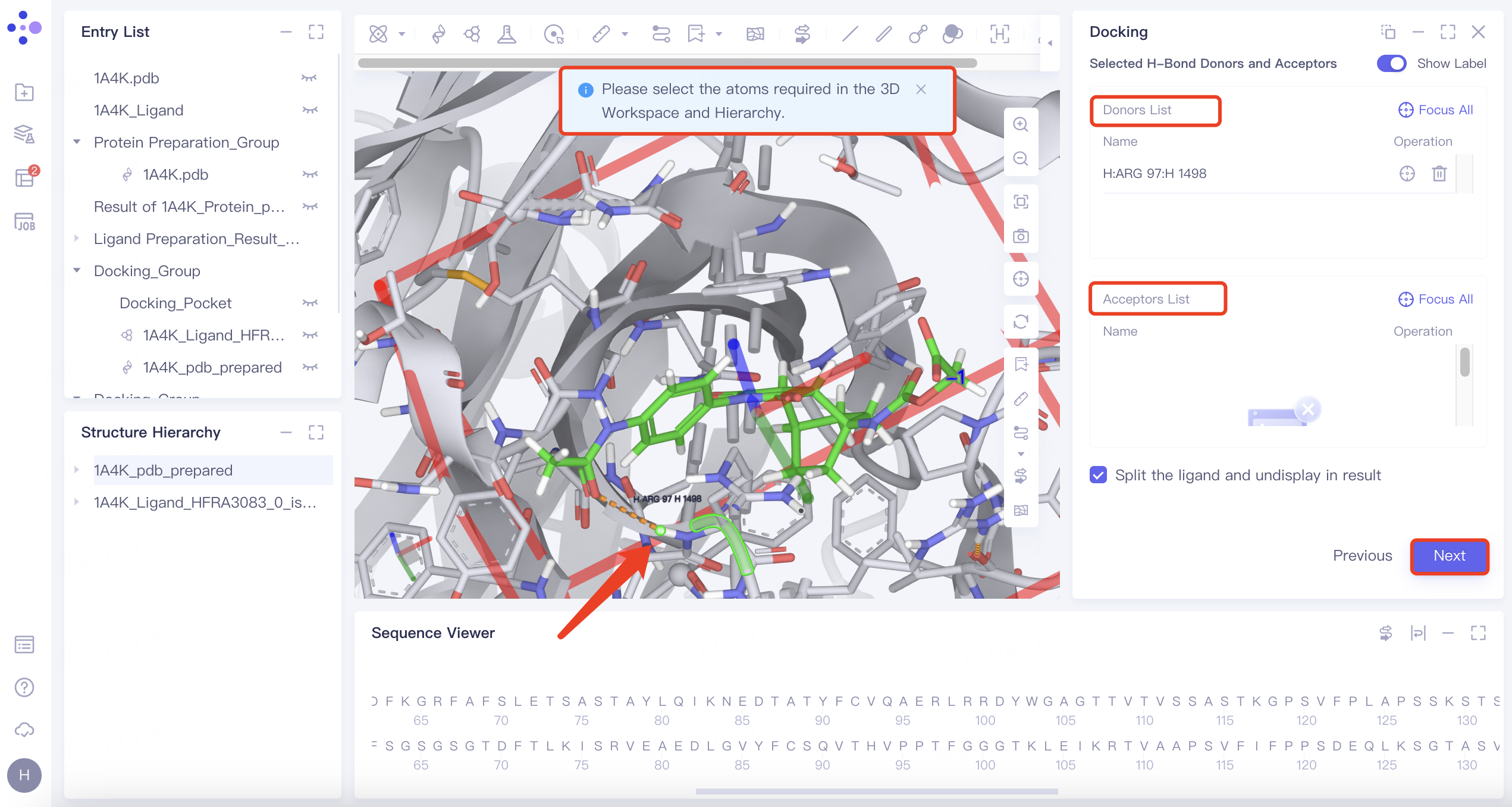 | 1)当配体与受体之间形成的某些氢键与分子活性相关时,可通过基于氢键限制的对接来筛选具有潜在活性的新分子 2)橙色虚线表示当前配体与受体之间存在的氢键相互作用 |
| Maximum Common Substructure | 1)点击“Maximum Common Substructure”按钮,点击“Confirm”; 2)选择参考配体(即与受体具有合适结合模式的配体),上传好的配体会自动进入力场检查,检查通过后Valid列显示“Valid”,同时Structure View窗口中展示分子2D图 | 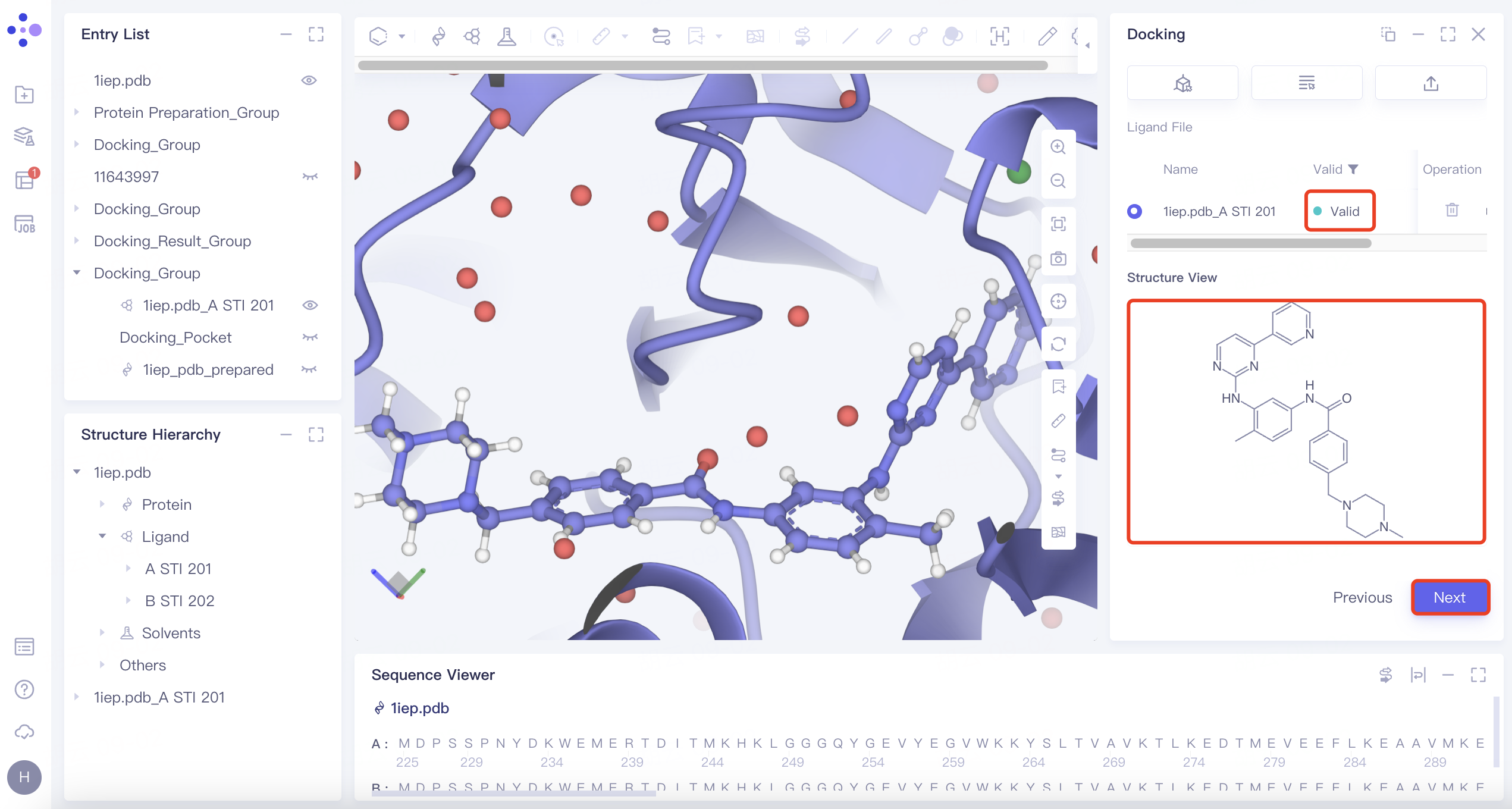 | 当您已知一个分子与受体的结合模式合适,需要从新的分子中筛选出与这种结合模式匹配的分子时,可以选择“Maximum Common Substructure” |
| Substructure | 1)点击“Substructure”按钮,点击“Confirm”; 2)选择参考配体(即具有特定子结构的配体),上传好的配体会自动进入力场检查,检查通过后Valid列显示“Valid”,同时Structure View窗口中展示分子2D图,选择需要的子结构后,点击“Save”保存该子结构 | 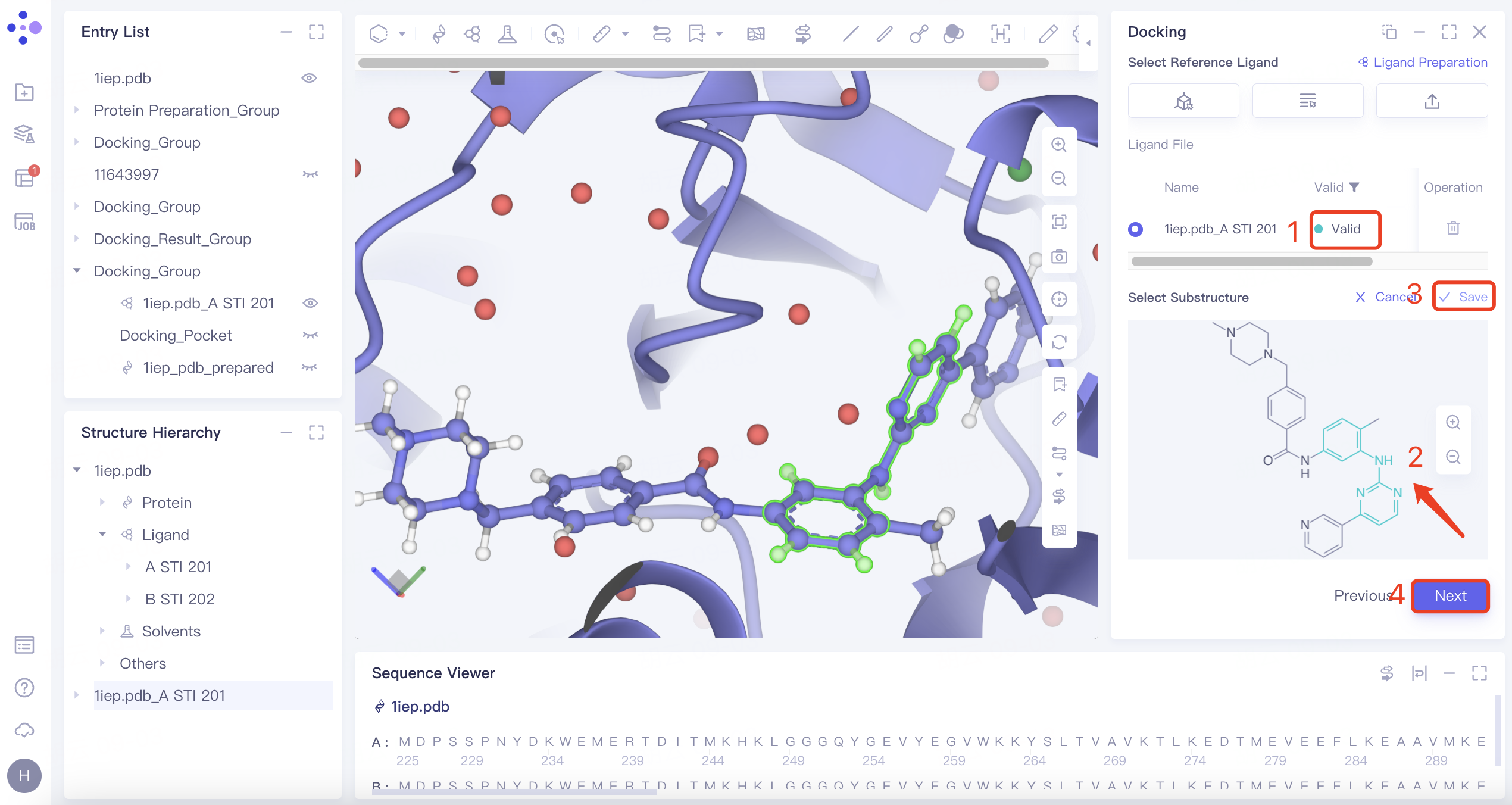 | Substructure选项提供了基于子结构筛选的分子对接,通过搜索出输入配体中包含特定子结构的配体,进一步与受体对接 |
| Shape | 1)点击“Shape”按钮,点击“Confirm”; 2)选择参考配体(即具有特定形状的配体),上传好的配体会自动进入力场检查,检查通过后Valid列显示“Valid”,通过输入在原分子体积上的扩大比例确定需要形成的形状,点击右侧“Sync to 3D”将该形状显示于3D Workspace窗口 | 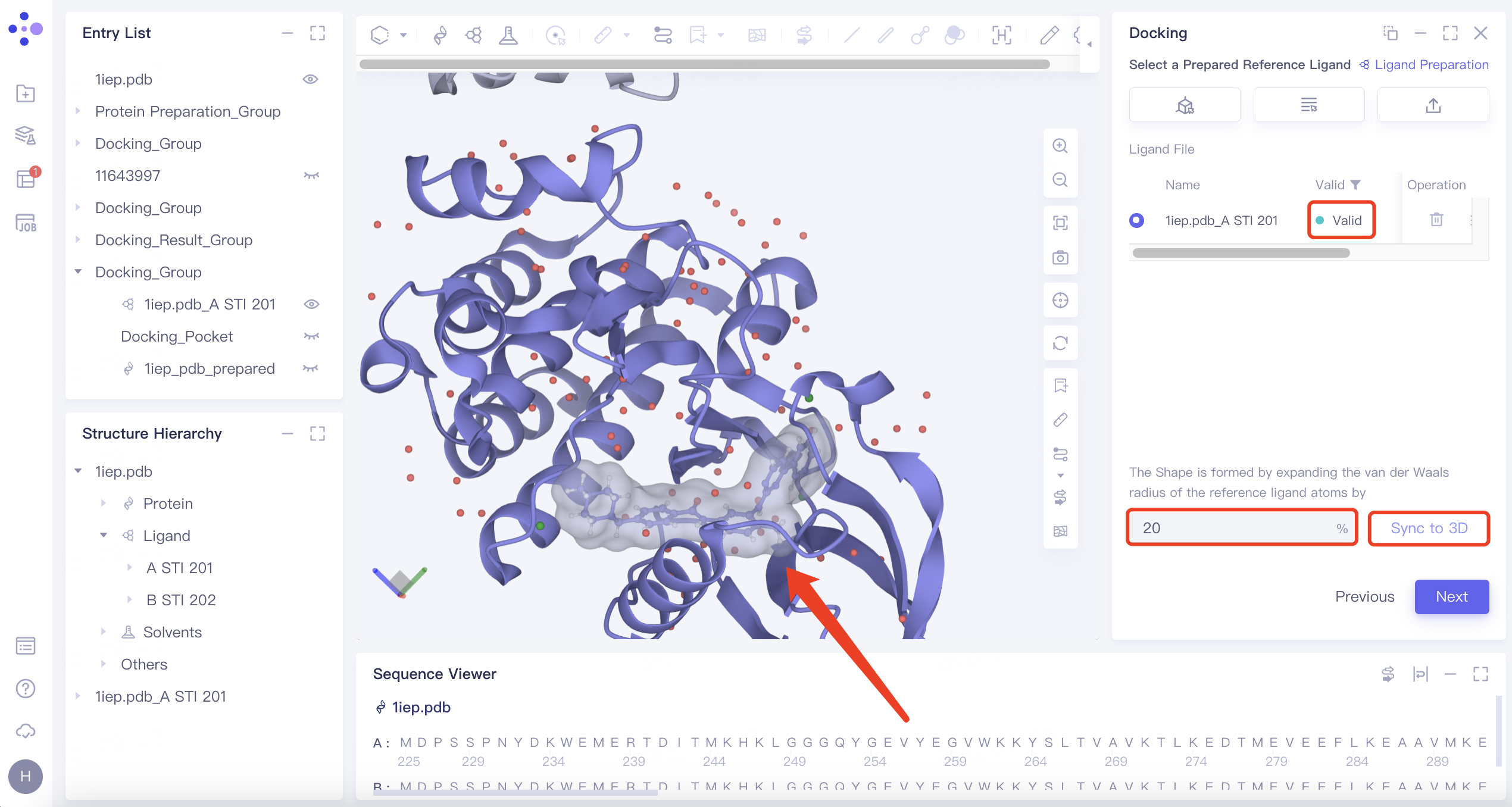 | 1)基于形状的虚拟筛选在骨架跃迁、生物异构体替代、虚拟库设计等方面具有重要意义,“Shape”选项提供了基于形状的虚拟筛选功 2)举例:比例设置为20%时,代表在原分子的体积上,扩大20%的范德华半径形成一个新的体积 |
1.6 Workflow Setting
勾选“Use Uni-Clip to retain ligands with the top 50% of rankings”,且将50%调整为70%;
勾选“1st:Fast Mode Docking”
Number of Results to Keep to 1st Docking处设置为Top Percent 20%;
其他参数默认;
勾选“2nd:Balance Mode Docking”
Number of Results to Keep to 1st Docking处设置为Top Percent 20%;
其他参数默认;
勾选“3rd:Detail Mode Docking”
Number of Results to Keep to 1st Docking处设置为Top Percent 20%;
其他参数默认;
勾选“Use Uni-Mol to optimize the resulting ligands' pose(s)”,优化对接构象;
勾选“Do rescoring”,打分函数选择“Gnina”;
MM PB/GBSA Setting保留默认参数;
Job Name处保留默认;
点击“Submit”提交任务。
| 参数 | 意义 | 注 |
| Uni-Clip | 一种基于对比学习算法的工具,根据训练得到的配体与靶标的关系,快速过滤构象不合适的配体 | 是一种粗略的过滤方式,建议保留70%的结果 |
| Fast/Balance/Detail Mode Docking | Fast:exhaustiveness 128 & max_step 20 | 三种不同的搜索模式,本质上设置了不同的分子对接计算复杂度,Detail对搜索空间的覆盖更全面,且准确性最高,其次为Balance,最后是Fast |
| Balanced:exhaustiveness 384 & max_step 40 | ||
| Detailed:exhaustiveness 512 & max_step 40 | ||
| Number of Results to Keep to 1st/2nd/3rd Docking | 保留结果数,可选All/Top Number/Top Percent | |
| Keep Multi Binding Poses for Each Ligand(每个配体生成多个构象) | 1)Number of Binding Pose,生成的构象个数,支持1~100; 2)Energy Range (kcal/mol):与最优结合模型相差的最大能量值,支持1~9 | |
| Scoring Function | 打分函数目前包括Vina、Vinardo、AutoDock4 | 一般选择Vina |
| Uni-Mol | 使用Uni-Mol优化对接后的pose | 本质上是一种基于机器学习方法的限制性对接 |
| Rescoring | 对对接构象进行重打分。重打分函数包括Vina(↓), Vinardo(↓), GNINA(↑), KarmaDock Affinity(↑), RTMScore(↑), Uni-Mol Affinity(↓), Uni-Score(↑) | 推荐使用Gnina |
注:MM PB/GBSA参数说明详见《MM PB/GBSA》。
2. 结果分析
2.1 任务入口
左侧通用菜单栏Job → 该任务生成了四个结果文件:7M94_protein_prep_Clip、7M94_protein_prep_VSW、7M94_protein_prep_Rescore和7M94_protein_prep_GBSA。
注:Virtual Screening Workflow最多产生如下五类子任务:
| Job类型 | 说明 |
| Ligand Prep | 当配体经过配体准备生成同分异构/构象,才产生该任务,当输入配体为平台提供的Database时则不产生该任务 |
| Uni-Clip | 勾选Use Uni-Clip to retain ligands with the top 50% of rankings时产生该任务 |
| VSW | 经过Fast、Balance和Detail搜索模式对接后的最终对接结果 |
| Docking Rescore | 对接构象的重打分结果,勾选 Do Rescoring时产生该任务 |
| MM PB/GBSA | 勾选MM PB/GBSA计算时产生该任务 |
2.2 Uni-Clip结果
7M94_protein_prep_Clip为使用Uni-Clip粗筛后的配体结果。
注:下载结果中包含一个表格文件,记录着Uni-Clip筛选出的配体结果,包含三列:Ligand ID、Ligand Name和Clip Score(Uni-Clip的打分值,越大越好)。
2.3 VSW结果展示
7M94_protein_prep_VSW为Uni-Clip粗筛后的配体通过三步分子对接后的结果,点击该任务Operation列中的Show显示该任务的结果。
对接结果默认按打分值进行排序,观察到Vina打分最低的pose的打分值为-12.724。
Docking Result表格说明:
| 列名 | 含义 | 注 | |
| Rank | 根据打分函数值的pose排序 | VSW类型任务默认按照Score Function排序,Rescoring类型任务默认按照Rescoring排序 | |
| Stars | 标星 | —— | |
| Ligand Name | 配体名称 | —— | |
| Score | 经过Fast、Balance和Detail搜索模式对接后pose的打分值 | 打分函数为最后一步搜索模式所选的打分函数 | |
| [Constrain Type] Score | 限制性对接的打分值 | 当Constrained Docking处对配体进行条件限制性对接时,产生该列数据 | |
| [Rescore Function] Rescore | Rescoring打分函数值 | (+)代表该打分函数值越高越好,(-��)表示值越低越好 | |
| Operation | Fix | 在3D Workspace中固定显示 | —— |
| 3D | 在3D Workspace中显示该配体 | —— | |
| Property | 查看性质信息 | —— | |
| Download | 下载该pose | 配体存为sdf格式 |
根据Rank排名,点击排名第一的pose右侧的3D按钮,将该pose展示于3D Workspace窗口中,并且可通过键盘上下键切换pose。合适的pose可打星。
配体以球棍模型显示,与配体具有相互作用的氨基酸残基以Line形式呈现,虚线表示相互作用,点击或悬停至虚线则于3D Workspce窗口左下角显示相互作用说明,不同的相互作用以不同的显色表示。
注:通过Show最多展示排名前3000的配体结果,全量数据可通过Job处的下载获取。
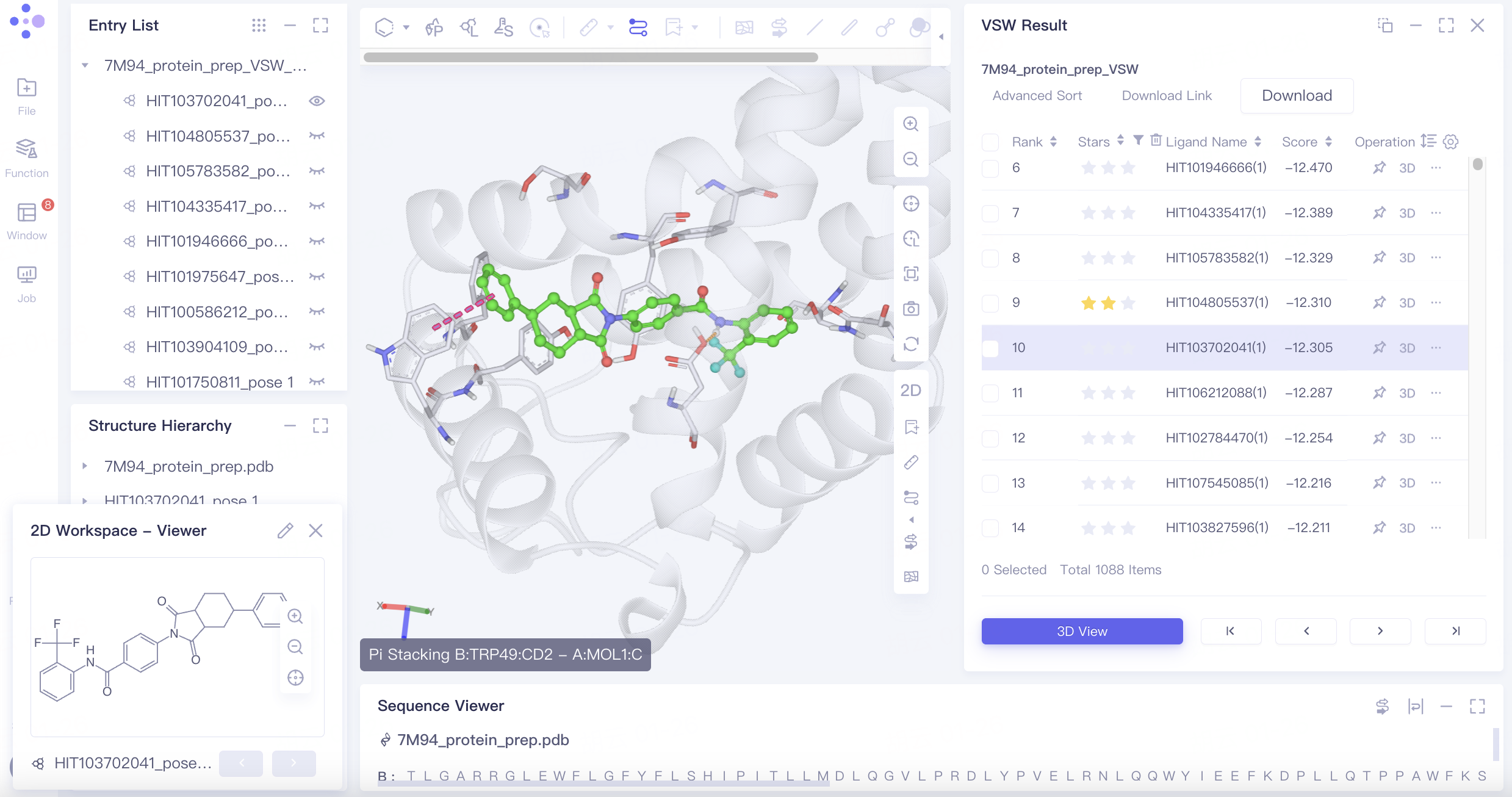
Tyr150与配体之间的Pi-Pi Stacking相互作用与配体的活性具有潜在相关性,因此期望筛选出与蛋白之间具有该相互作用的pose,可通过如下操作实现:
点击“Advance Sort”,在弹出的界面中找到IFP Filter,热图中作如下选择,点击“Confirm”,筛选后的结果重新排列于结果表格中。
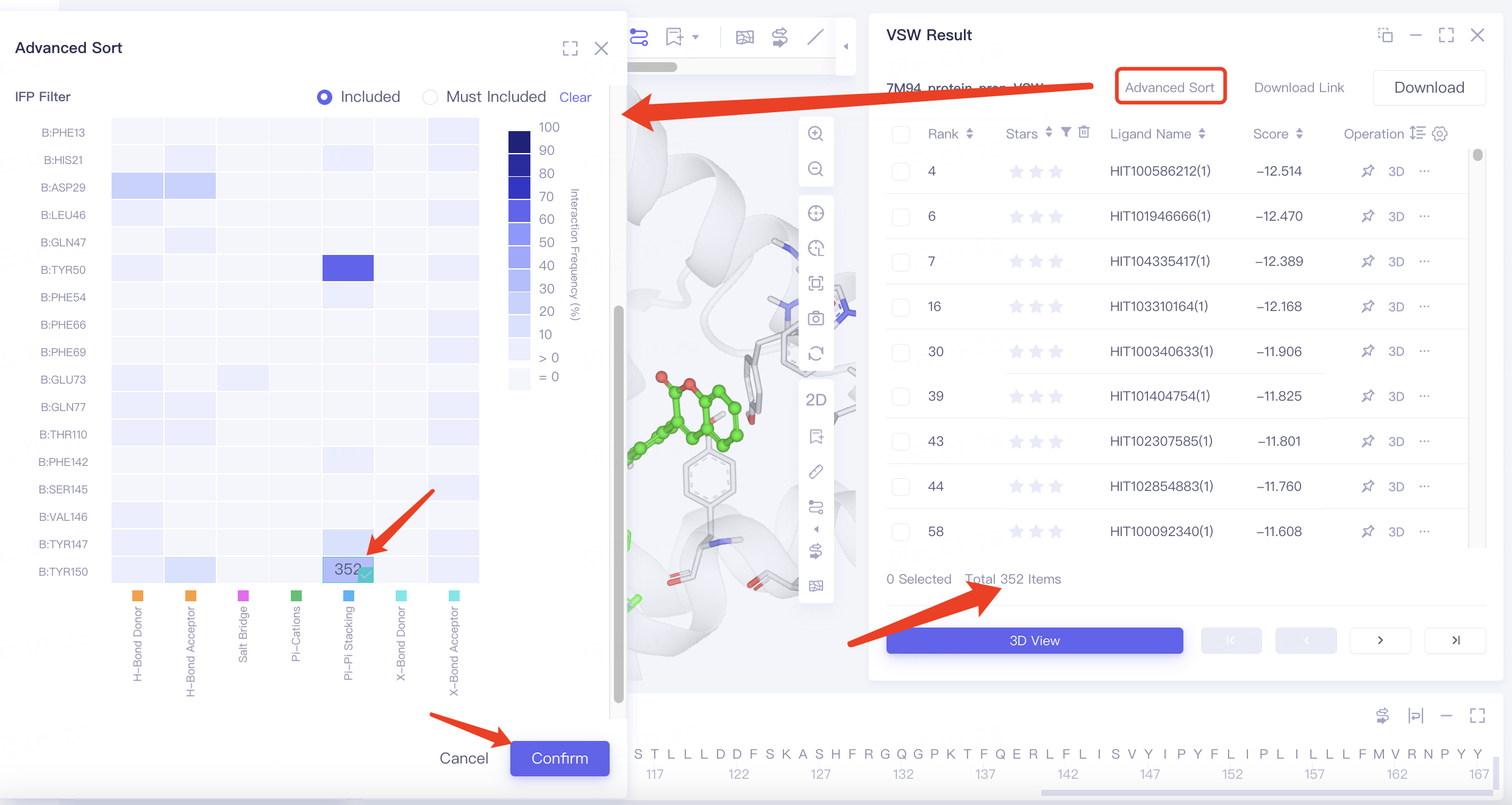
注:Advance Sort可用来筛选pose,包含如下方式:
-
Result Filter:根据Star、Score以及Rescore值的范围对Docking结果进行过滤;
-
Result Rank:根据Ligand Name、Star、Score以及Rescore对Docking结果进行排序;
-
IFP Filter:根据对接结果中配体与�蛋白之间的相互作用过滤Docking结果;
-
Include/Must Include:Include代表当Docking结果包含所选的任意一种相互作用时,即保留该结果;Must Include代表Docking结果必须包含所选的所有的相互作用时,才保留该结果。
-
热图:横轴为相互作用类型,纵轴为残基及其位数,图中的颜色深浅代表Docking结果中,配体与残基之间出现相互作用的频率。悬停至热图的小框中,显示的数字代表Docking结果中,存在该相互作用的结果数。
-
2.4 Rescoring结果展示
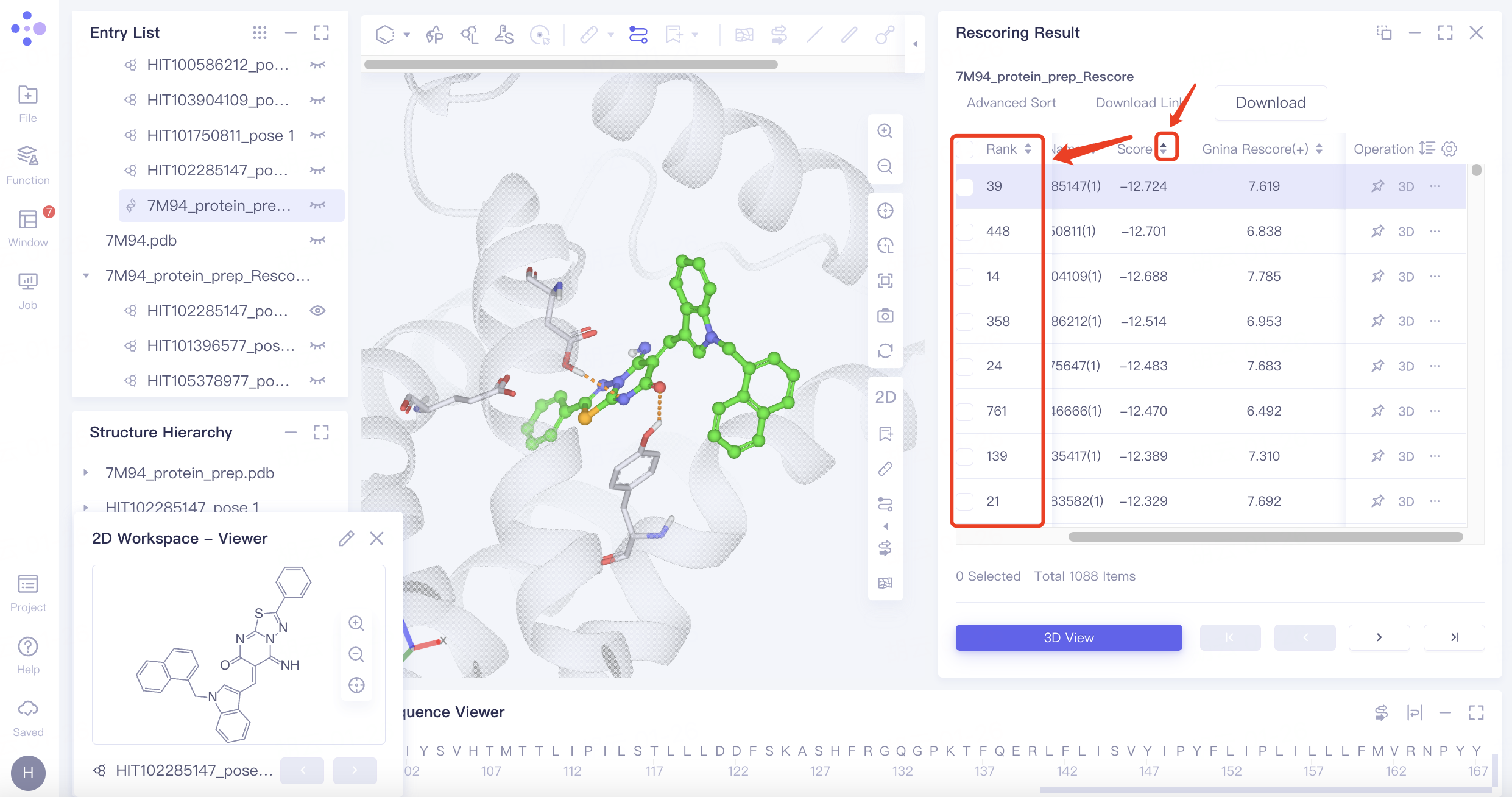
7M94_protein_prep_Rescore是该任务的Rescoring结果,与7M94_protein_prep_VSW的差别在于结果列表中具有“Gnina Rescore(+)”列,并且结果表格默认按照“Gnina Rescore(+)”列打分值进行排序。
Rescoring提供了更加多样的打分函数,且由于打分方式的差异,虚筛结果会有较大差异。本教程的案例,当根据“Score”列进行重新排序后,结果如下,在这批分子中,Score(Vina)打分第一的pose使用Gnina打分仅排第39名。结合Rescoring,您可以更加全面地筛选出所需配体。
2.5 MM PB/GBSA结果
MM PB/GBSA是计算配体与蛋白质受体之间的结合自由能的一个工具,它比大多数分子对接打分函数更准确。
7M94_protein_prep_GBSA是该任务的MM PB/GBSA计算结果,计算得到的结合自由能结果顺序如下图所示。点击右侧Operation下的3D,可将该pose展示于3D Workspace窗口中,根据ΔG Total值及分子构象,您可进一步筛选分子。
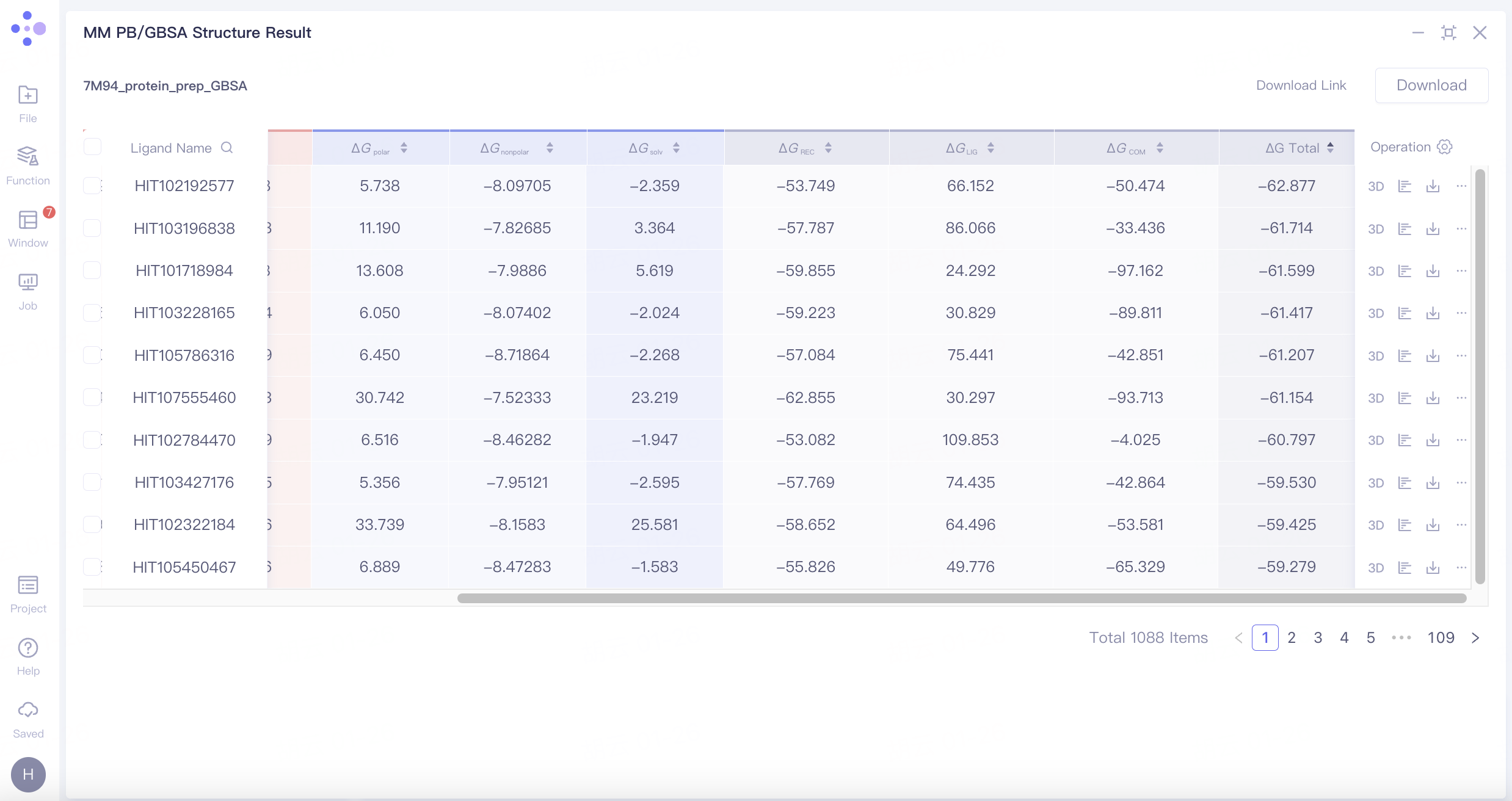
MM PB/GBSA计算结果展示:
提供溶剂态、气态和总和3种维度的ΔG数据:
注:详见《MM PB/GBSA》。
3. 参考文献
[1] Yu Y, Cai C, Wang J, et al. Uni-dock: Gpu-accelerated docking enables ultralarge virtual screening[J]. Journal of Chemical Theory and Computation, 2023, 19(11): 3336-3345.
[2] Alon A, Lyu J, Braz J M, et al. Structures of the σ2 receptor enable docking for bioactive ligand discovery[J]. Nature, 2021, 600(7890): 759-764.