Aquasite
Introduction
Water molecules are widely distributed in drug target proteins and play an important role in the interaction between drugs and target proteins. In drug design, it is an effective way to optimize the structure of compounds by replacing the key water molecules around the drug molecules and forming interactions between the drug molecules and water molecules. In recent years, a series of cases have emerged to improve the binding activity of compounds, enhance the selectivity of compounds and improve their pharmacokinetic properties by reasonably replacing water molecules.
Trujillo [1] et al. Designed a series of hH-PGDS inhibitor lead compounds (compounds 9, 11, 13, 14) by replacing two key water molecules (called primary water and auxiliary water in the literature). The changing trend of the affinity of the compounds was explored. The experimental results of Trujillo et al. Were as follows: compound 11 was obtained by replacing auxiliary water, and its activity was 2 ~ 3 times lower than that of compound 9; compounds 13 and 14 were obtained by replacing primary water, and their activity was 100 times lower than that of compound 9. This indicates that primary water is thermodynamically more stable than auxiliary water.
In this tutorial, you will learn to use the Aquasite module of the Hermite ® platform, focusing on the two key water molecules (primary water, auxiliary water, In this tutorial, the position prediction accuracy and binding free energy of water 1 and water 2 are calculated respectively, and the stability and importance of water 1 and water 2 are evaluated by calculating the free energy difference of water molecules, which provides guidance for drug molecular design.
The template used to design the compound in this document:
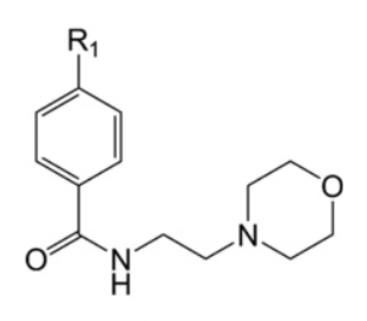
| Compound | R1 | hH-PGDS IC50(nM) | PDB ID |
| 9 | 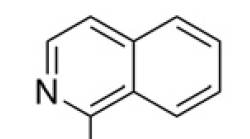 | 2.34 | 4EE0 |
| 11 | 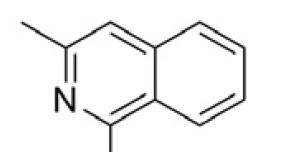 | 8.26 | 4EDZ |
| 13 | 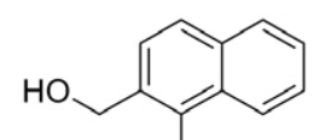 | 1480 | 4EDY |
| 14 | 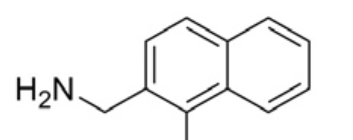 | 845 | 4EC0 |
1. Create a project and import the structure
1.1 Log in to the system
- Login address: https://hermite.dp.tech

1.2 Create the project
- Once in the system, create a new project �“hH-PGDS”
1.3 Import Protein Structure
- Left general menu bar Menu → File → Get PDB
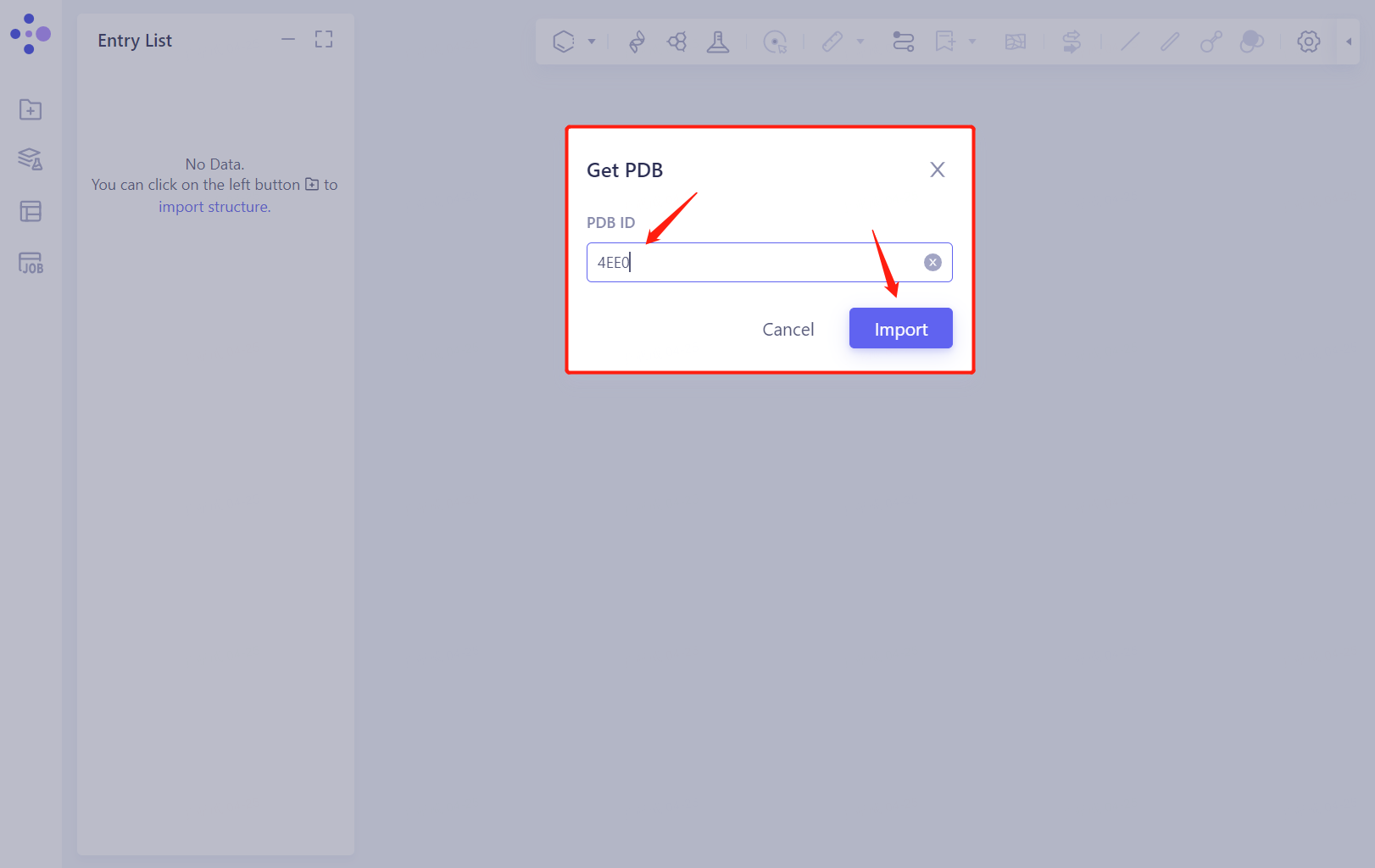
- Enter PDB ID: 4EE0 and import the protein
2. System preparation
2.1 Treatment system
-
In the Structure Hierarchy:
-
Select “Protein” Chain A and delete it;
-
Select “Ligand” A 0O4 202, A GSF 203, B GSF 202, delete ”;
-
Select “Others” Metal/Ions and delete it;
-
Select “Solvents” all A HOH water molecules, delete, keep B HOH water molecules
-
Note: Batch deletion can be performed by checking (click the start point, press and hold Shift, and click the end point to check).
-
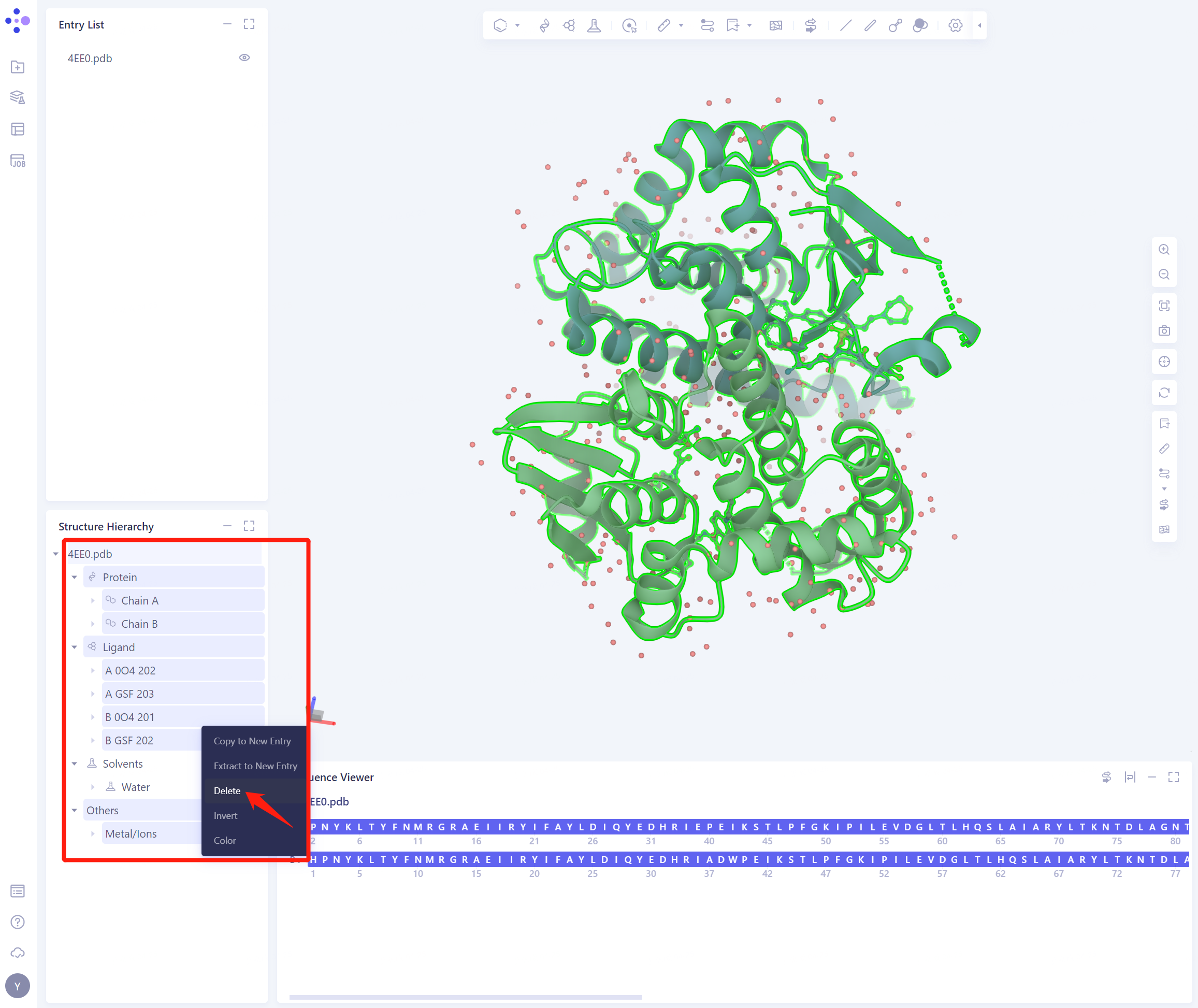
- After deletion, the protein structure is as shown in the figure:
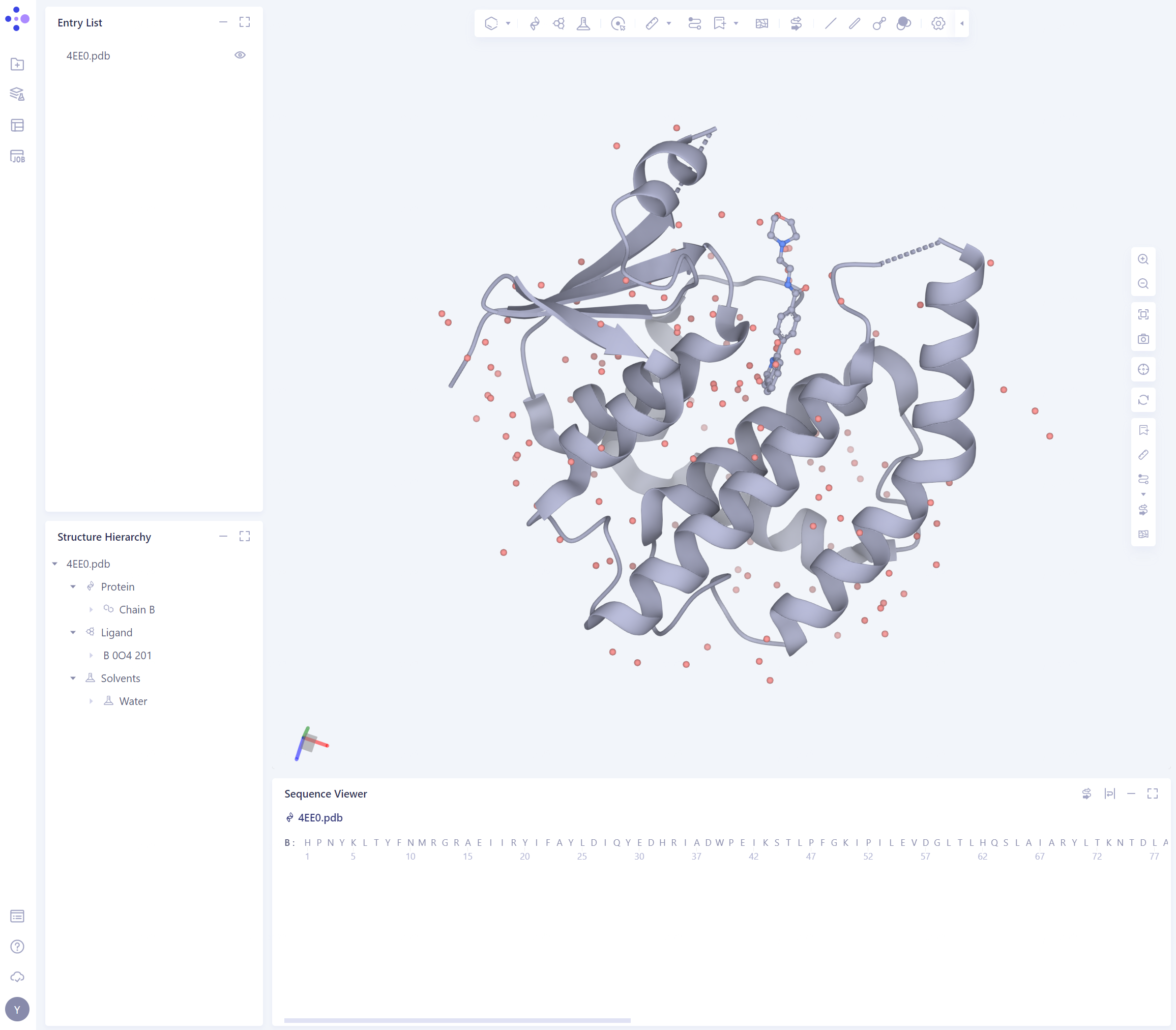
2.2. Prepare protein structure
2.2.1 Select Structure
- Left general menu bar Function → Structure Modeling → Protein Preparation
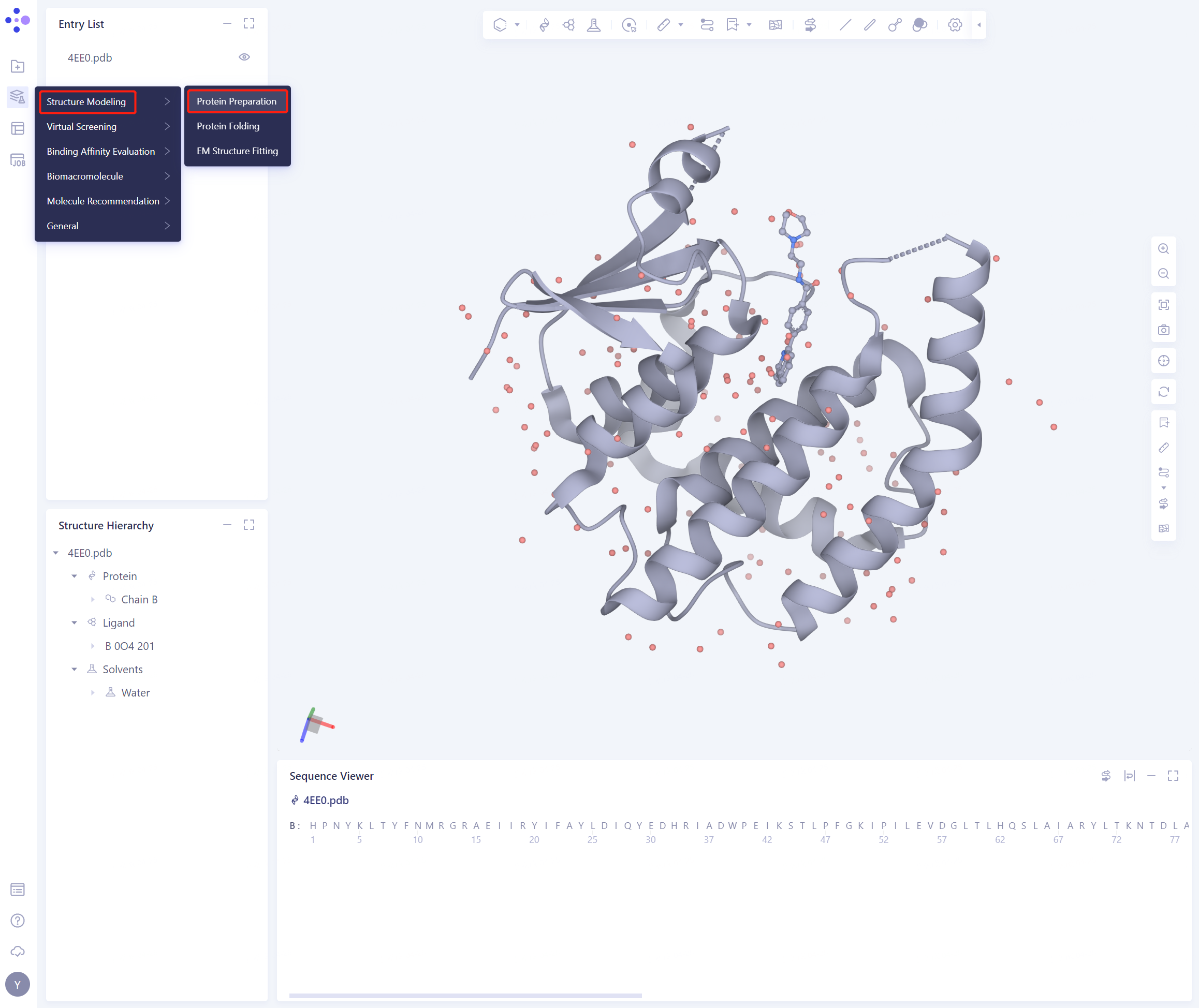
- Select Structure from 3D Works pace, taking care not to select small ligand molecules
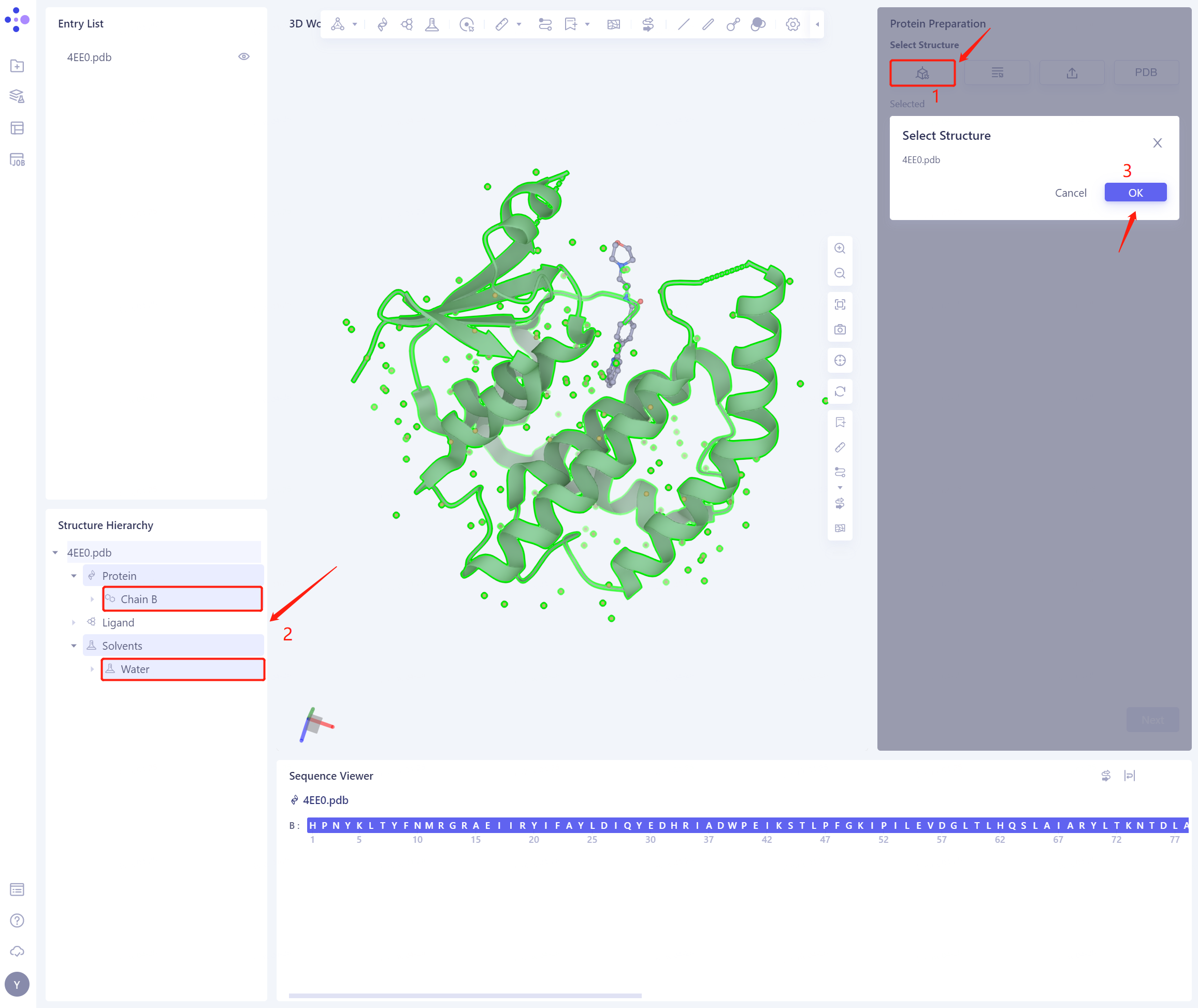
- 4EE0.pdb is loaded into the Protein Preparation parameter setting panel, and click Next
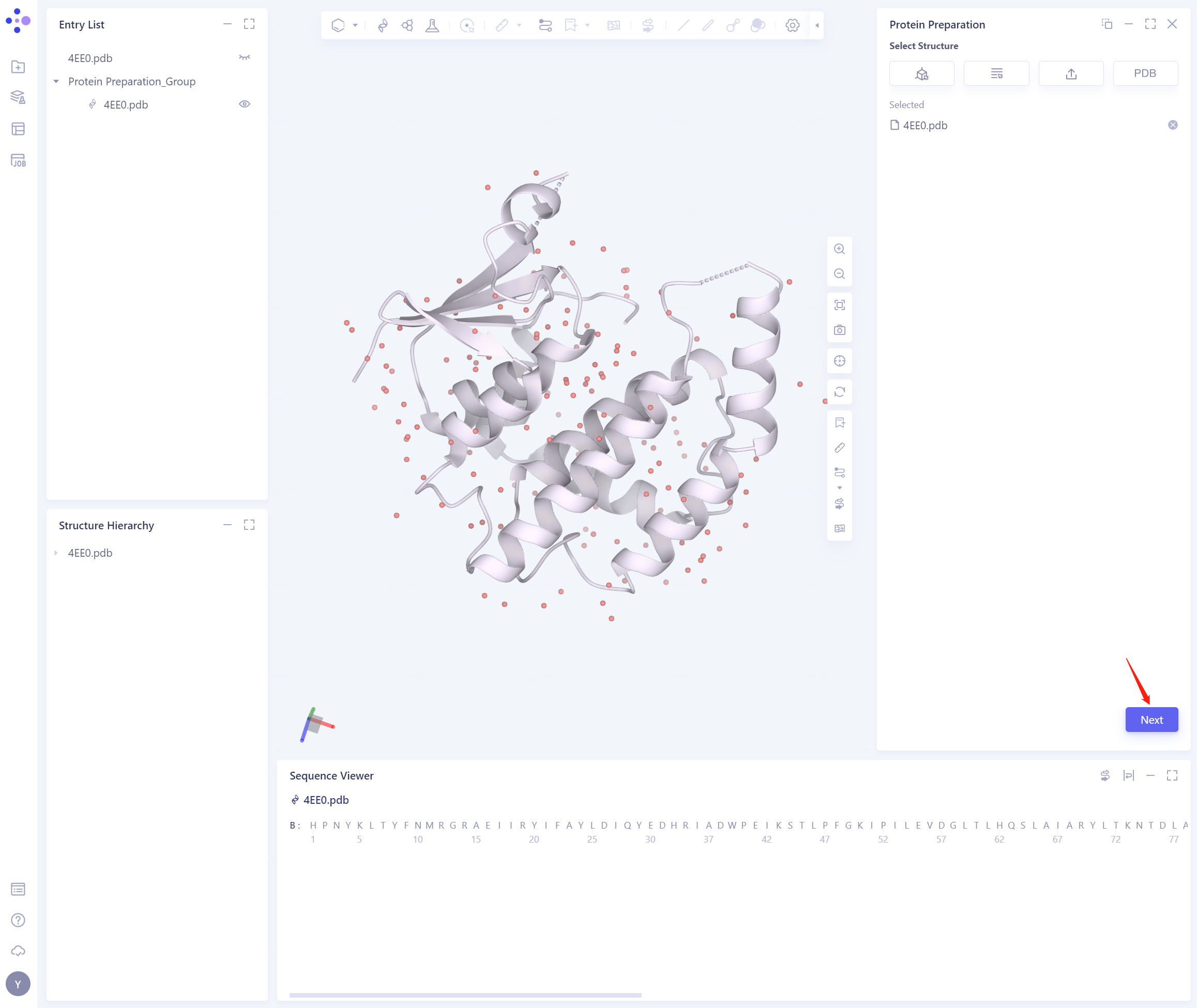
- Select the position of B-HOH (No.416) water molecule and click Next
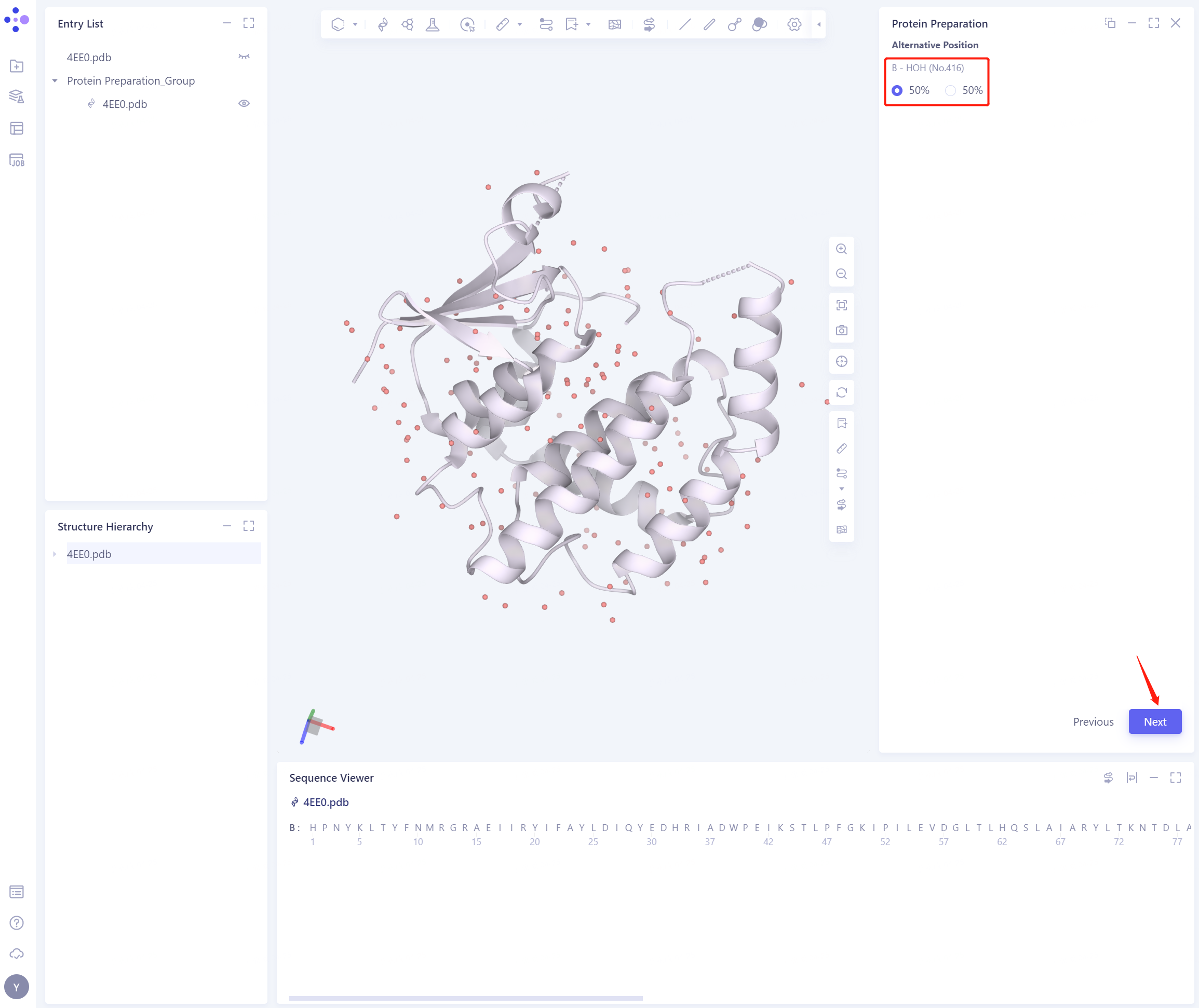
2.2.2 Select Polymer、Other Groups to Keep:
- Select the B-chain protein and the water molecule, and click Next
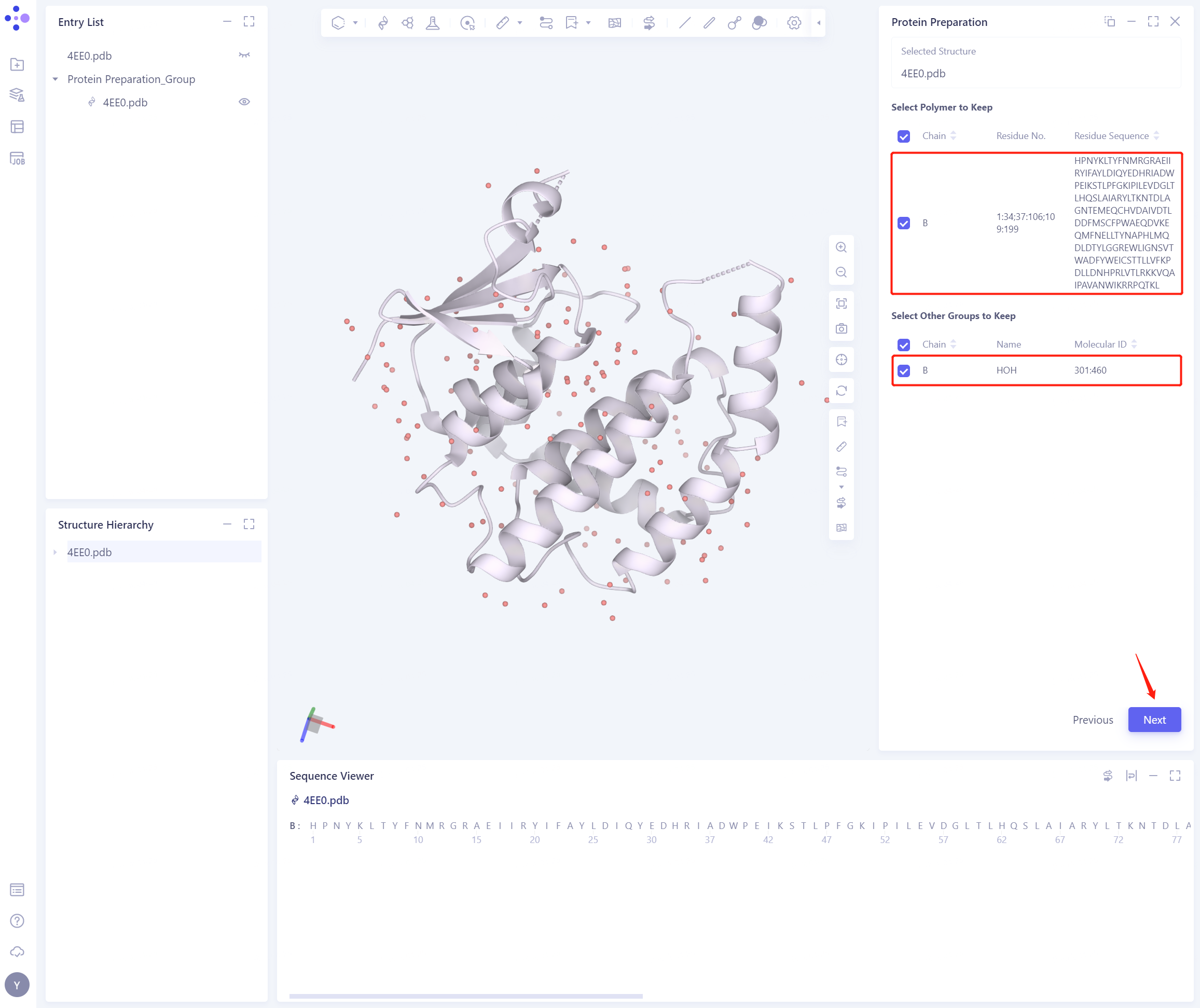
2.2.3 Select Missing Residues to Repair
- Check Missing Residues in this case
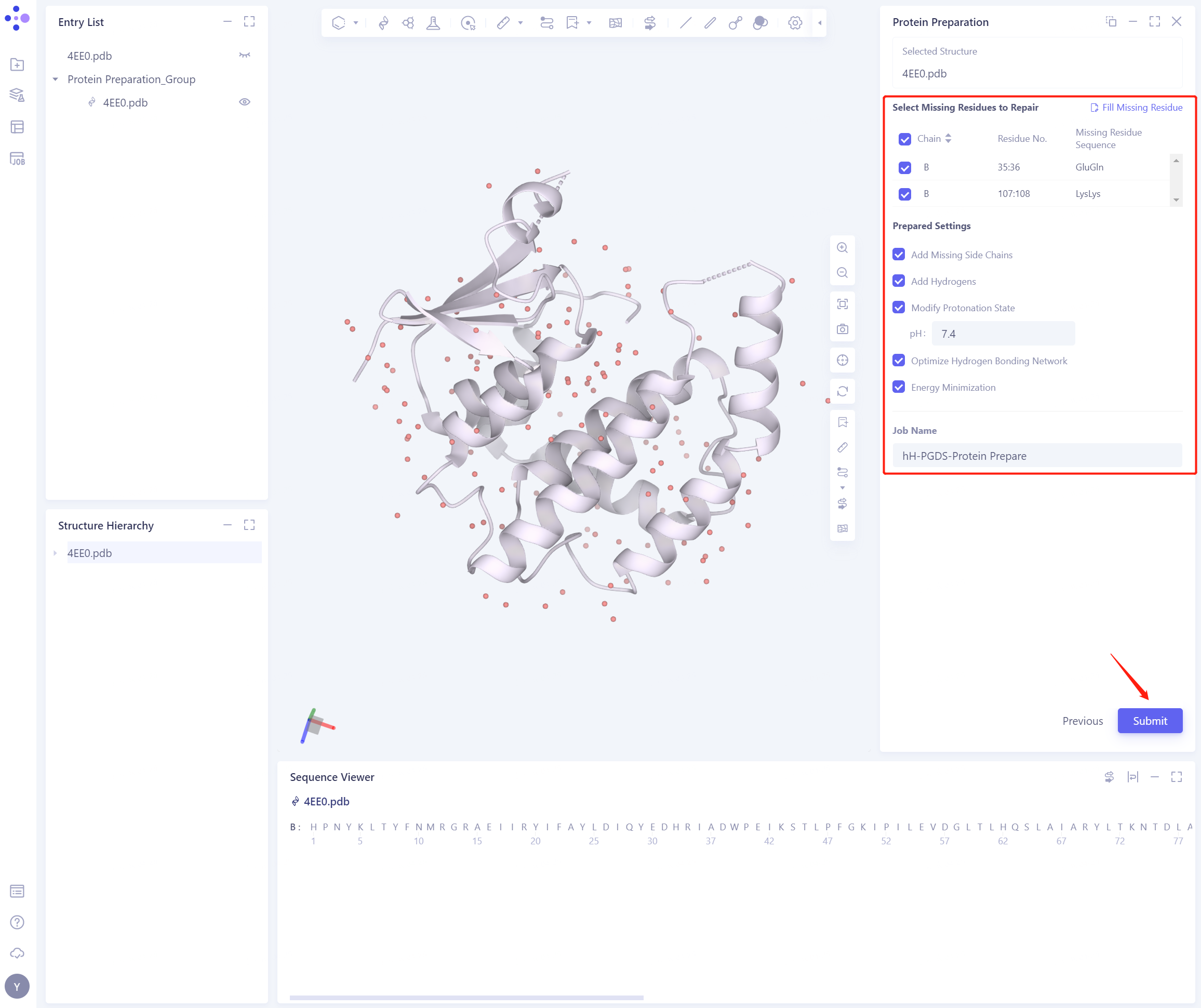
2.2.4 Prepared Settings
- Set the parameters according to the following figure.
2.2.5 Name the Job and submit the task
- Name the Job as “hH-PGDS-Protein Prepare” and click “Submmit” to submit the job
2.2.6 View Protein Preparation Result
- Protein Preparation computing tasks are generally completed in a dozen seconds to a few minutes. After the task is completed, click Jobs to view the corresponding task;
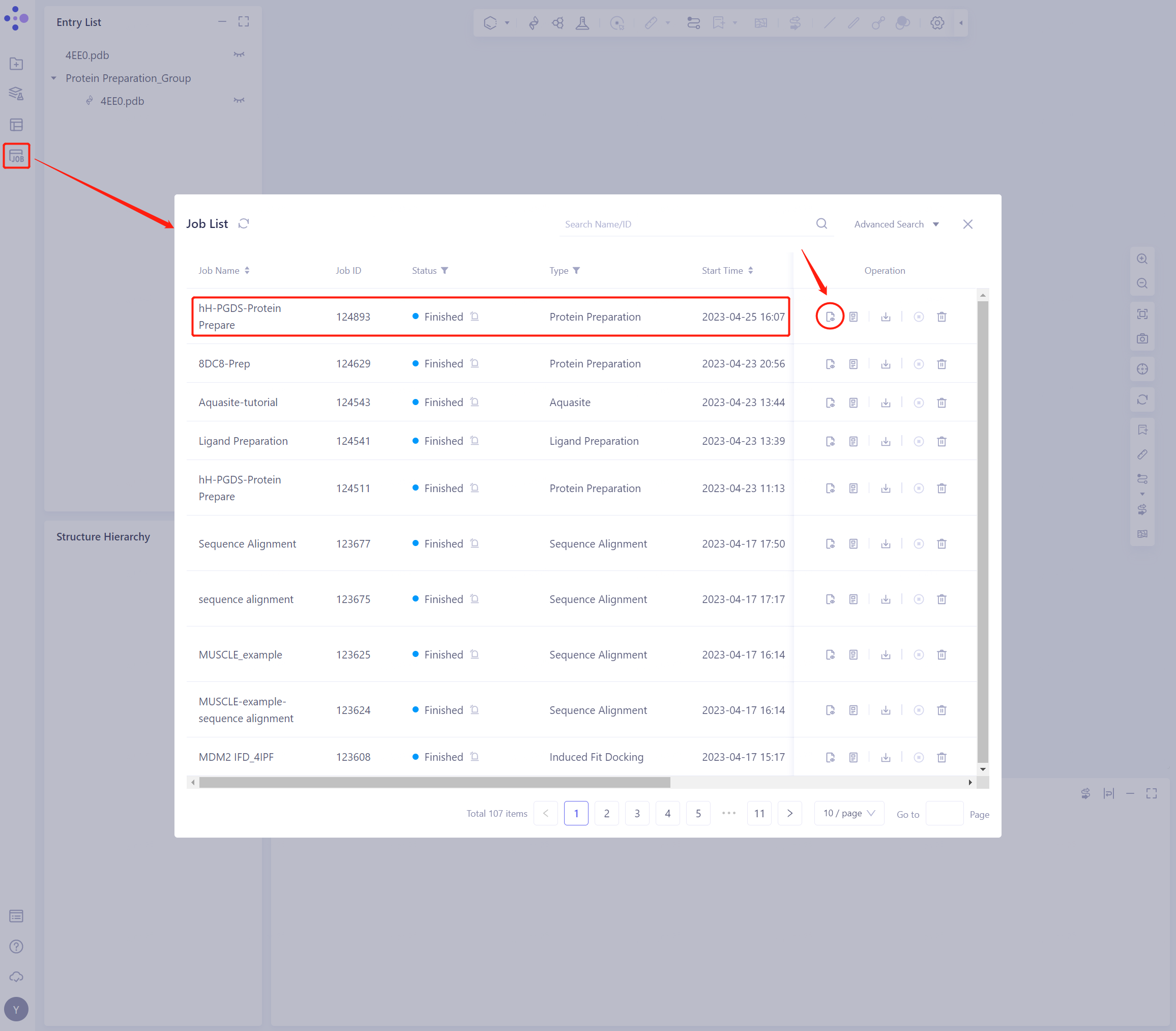
- Click the “show” button to display the prepared protein structure in 3D Workspace
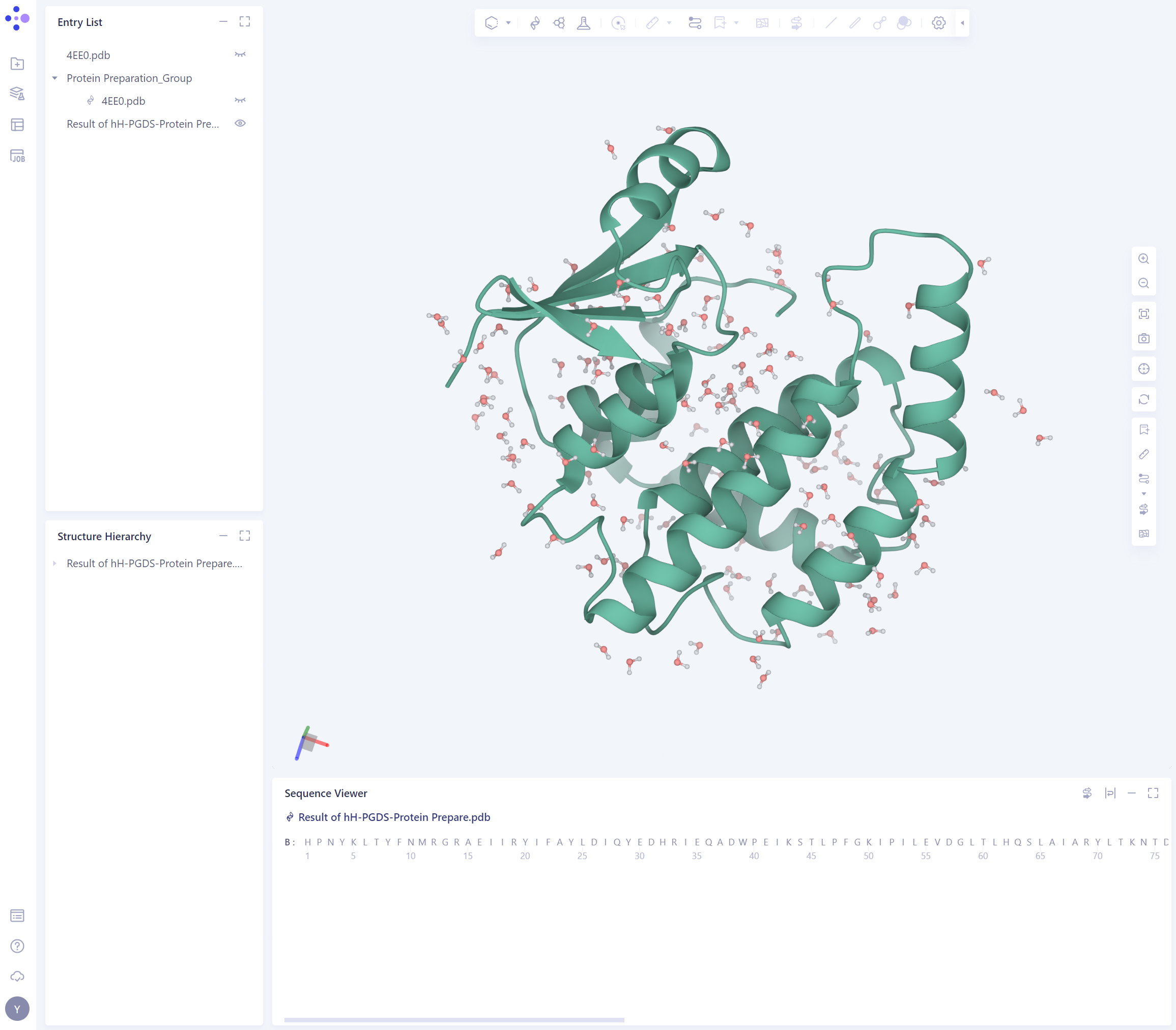
2.3 Prepare the Ligand Structure
2.3.1 Select Structure
- Left general menu bar Function → Virtual Screening → Ligand Preparation
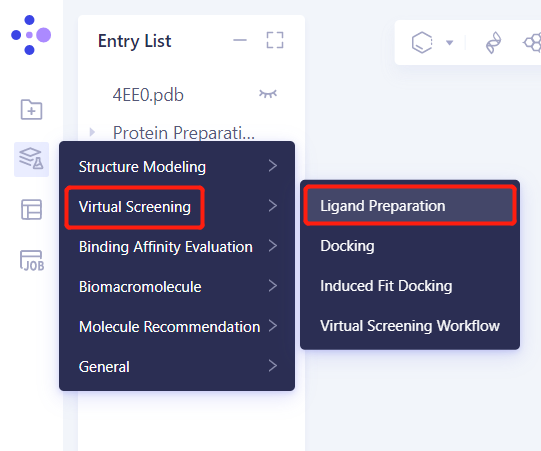
- Select Ligands from 3D
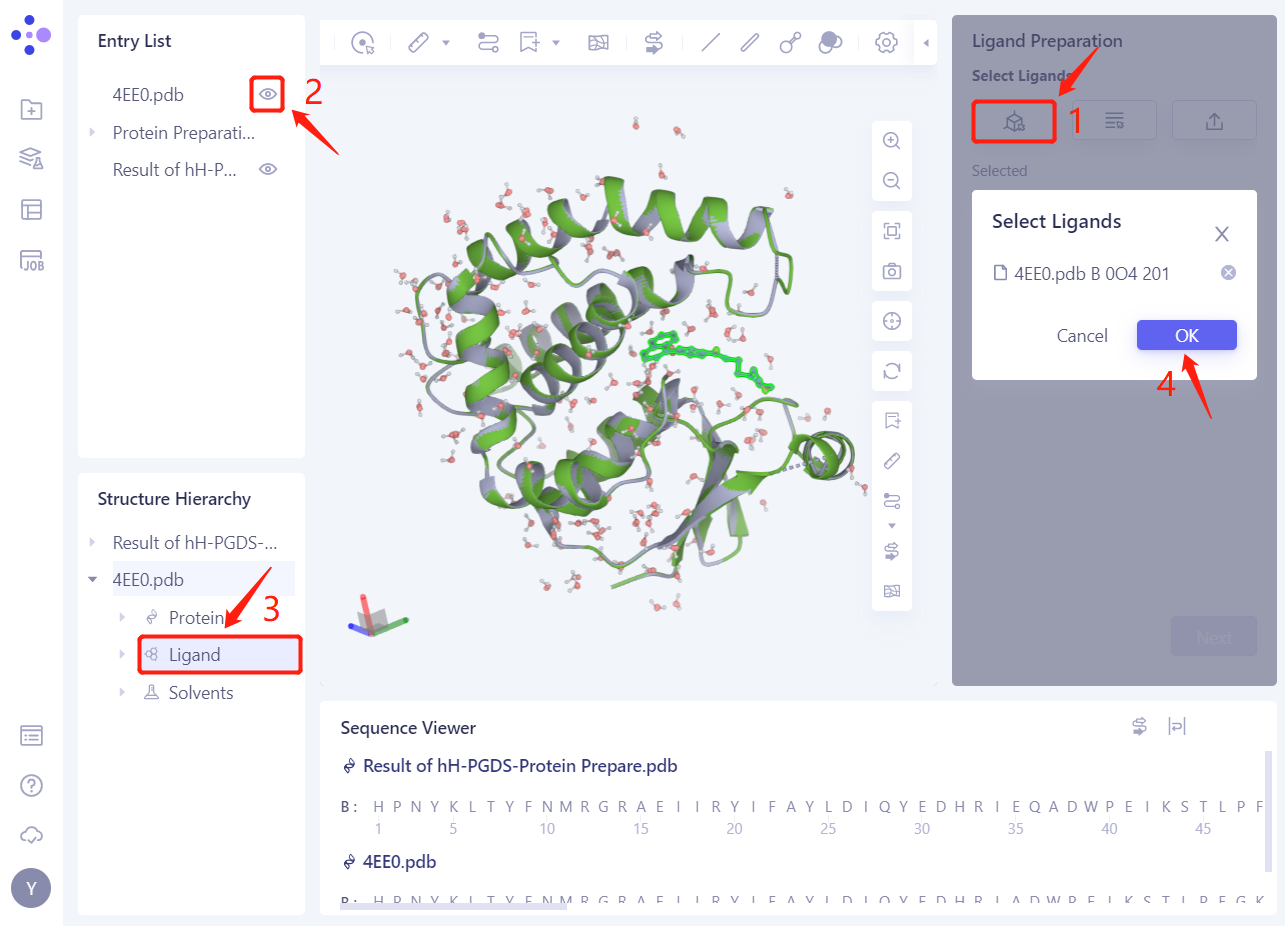
- Prompt whether to repair the possible missing bond information in Ligand, click Yes
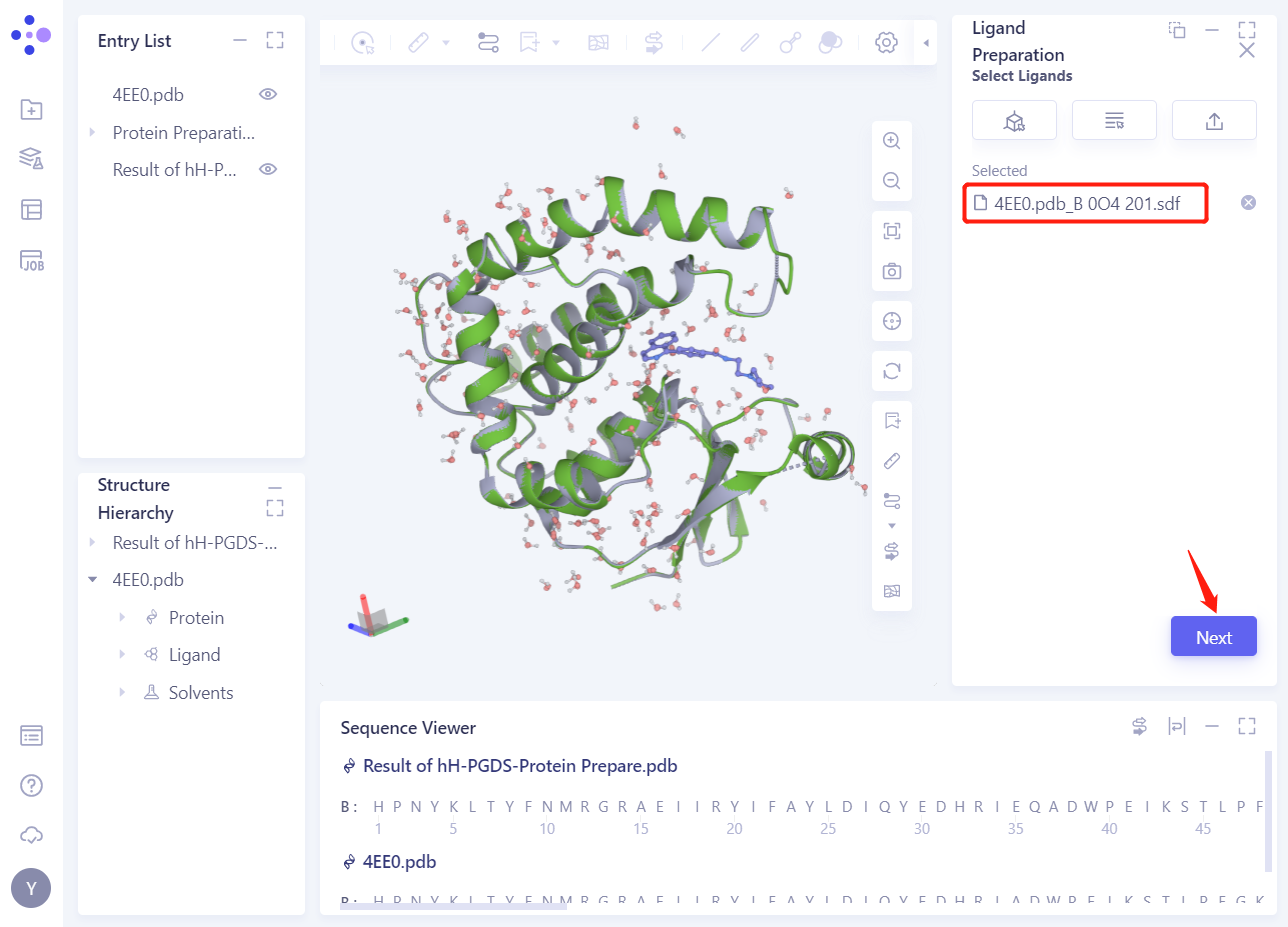
- 4EE0. PDB _ B 0O4201 The ligand is loaded into the Ligand Preparation parameter setting panel. Click Next.
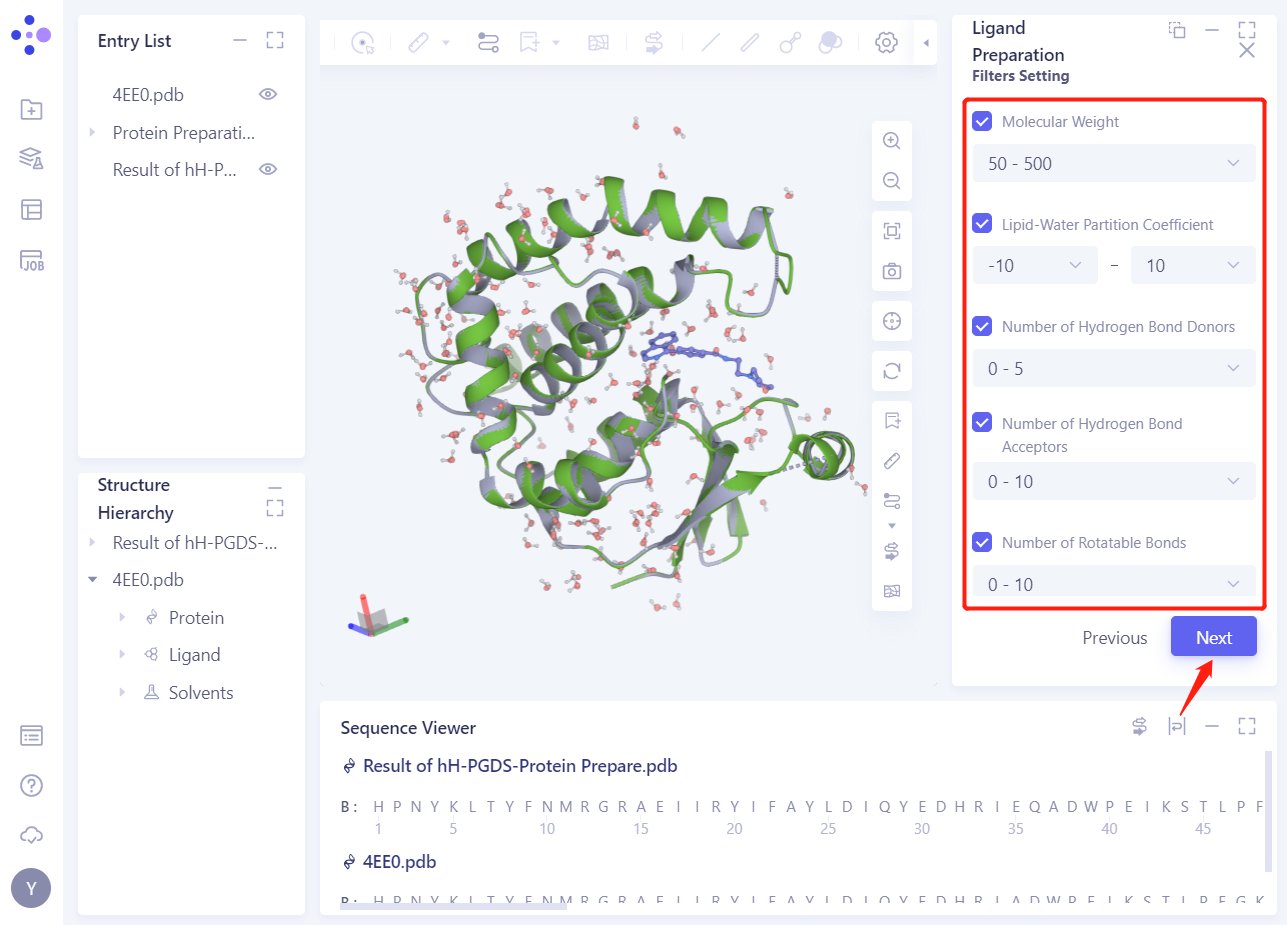
2.3.2 Filters Setting
- Select the default parameter settings and click Next
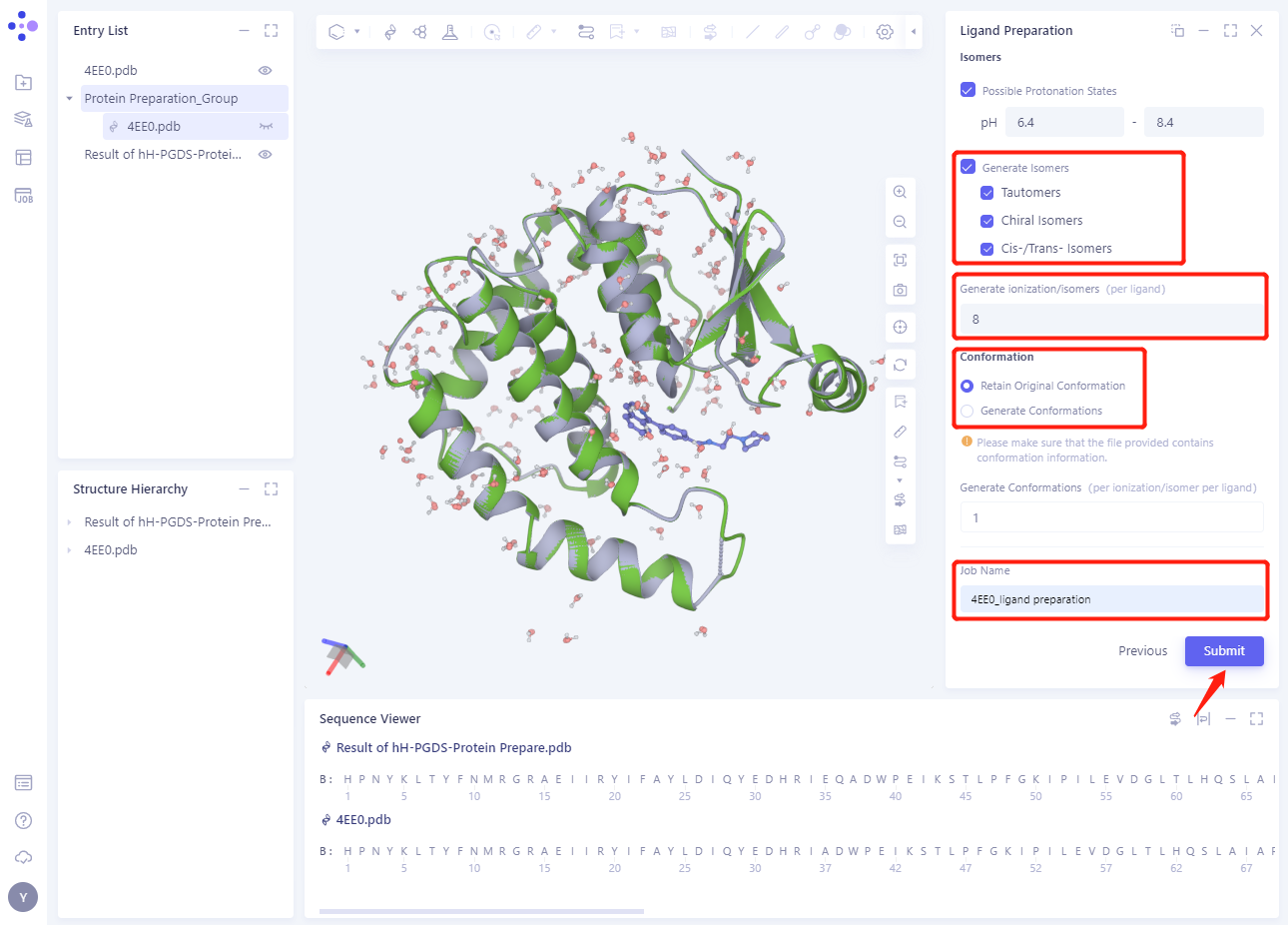
2.3.3 Isomers
- Set the Ligand Preparation parameters according to the red box in the figure below (see the Ligand Preparation module for details).
2.3.4 Viewing Ligand Preparation Result
- After the task is completed, click the show button of the task to view the corresponding task result
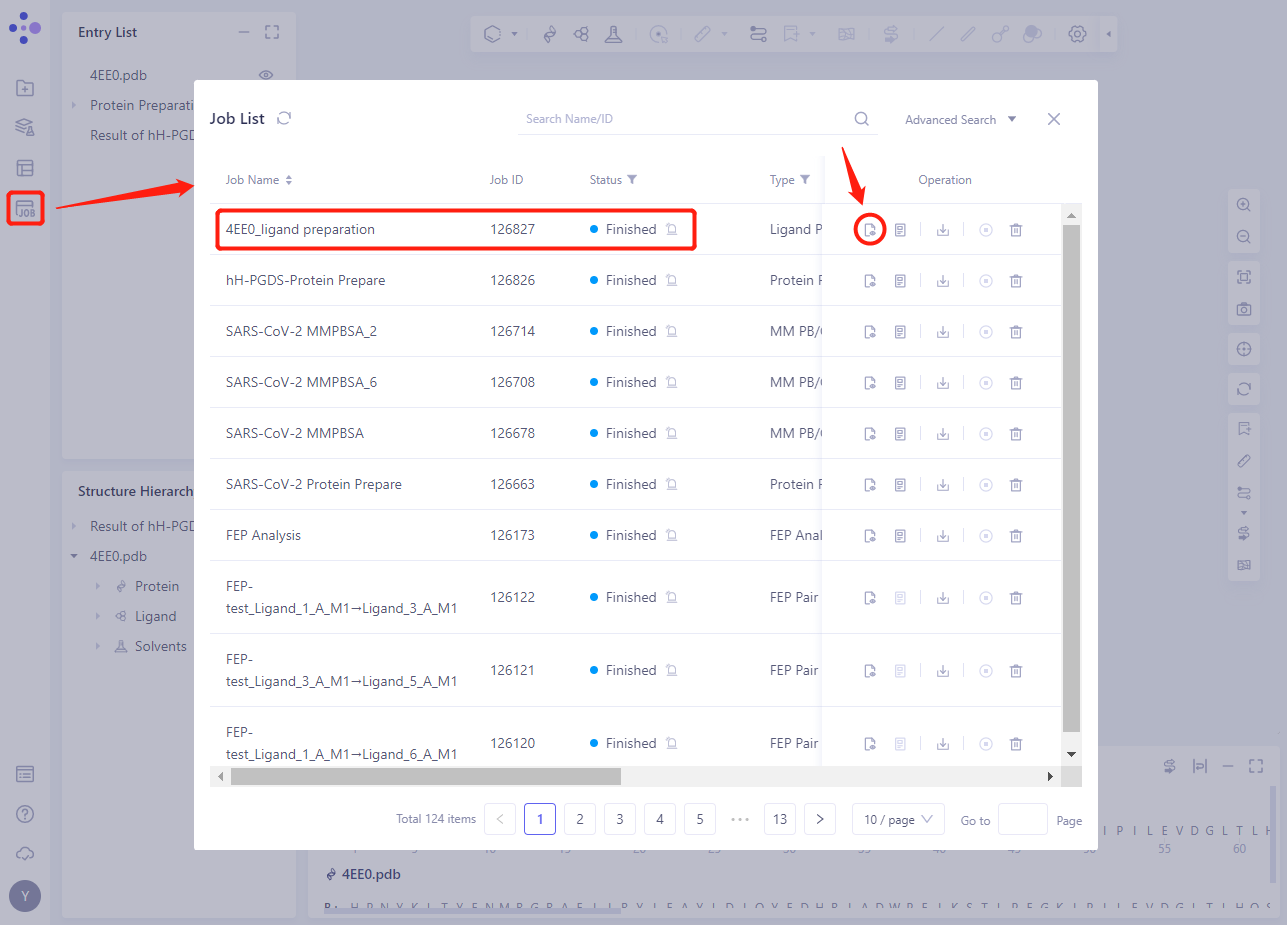
- Click the 3D button to display the processed ligand structure in 3D Workspace
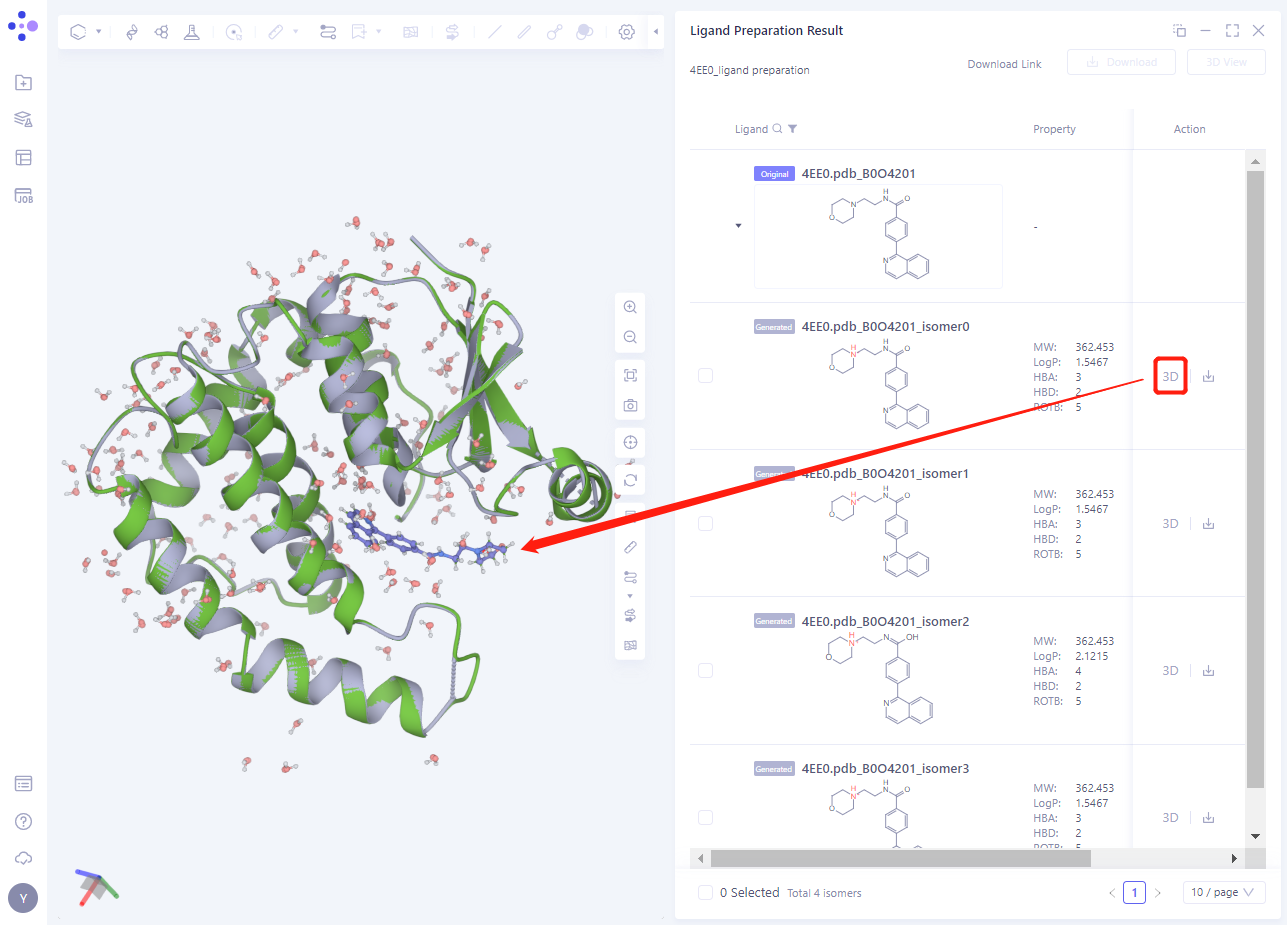
3. Create an Aquasite task
3.1 Left general menu bar Menu → Function → General → Aquasite
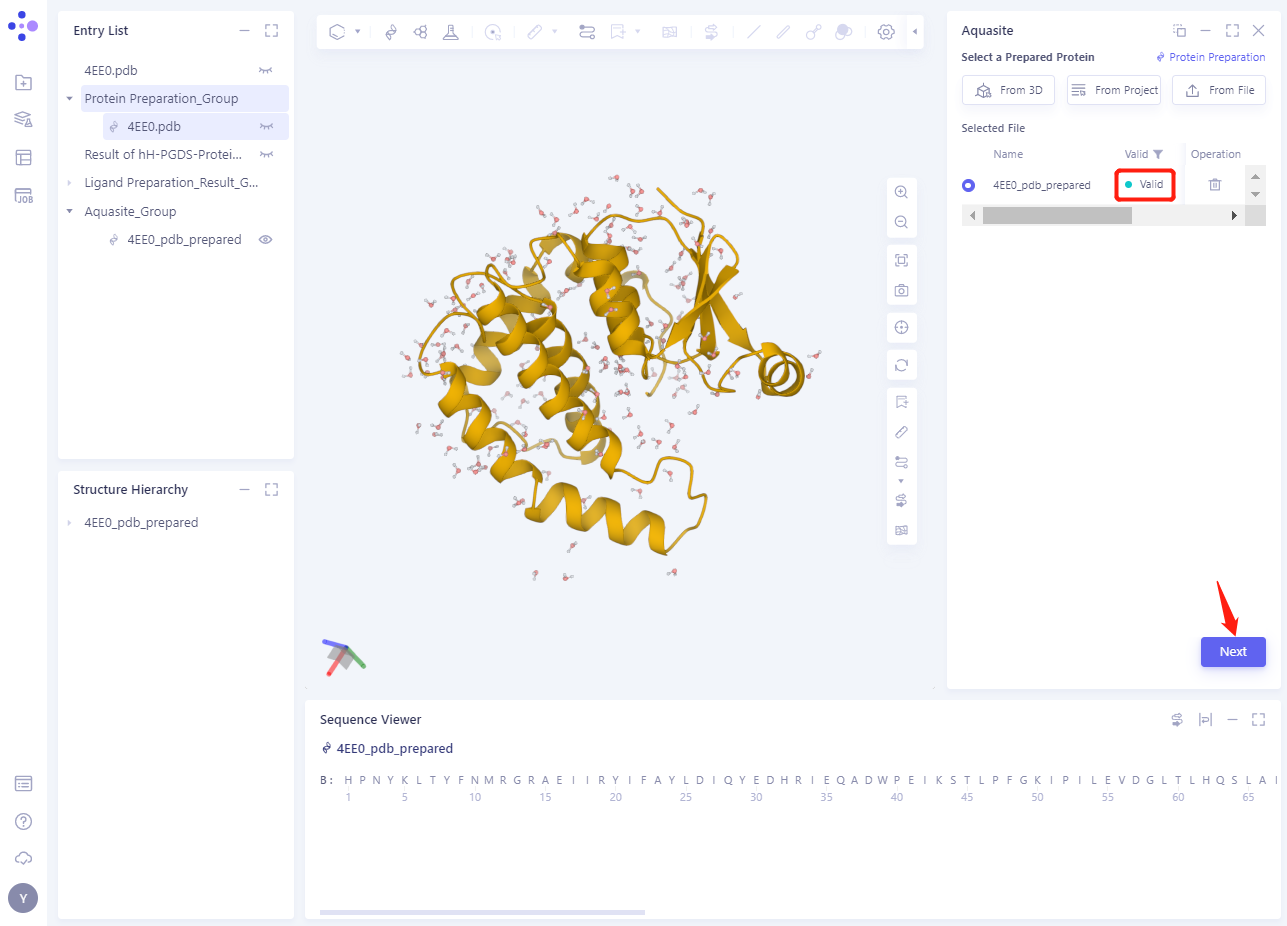
3.2 Select a Prepared Protein From Project
- Select “4EE0 _ PDB _ prepared” “as the prepared protein structure and click OK
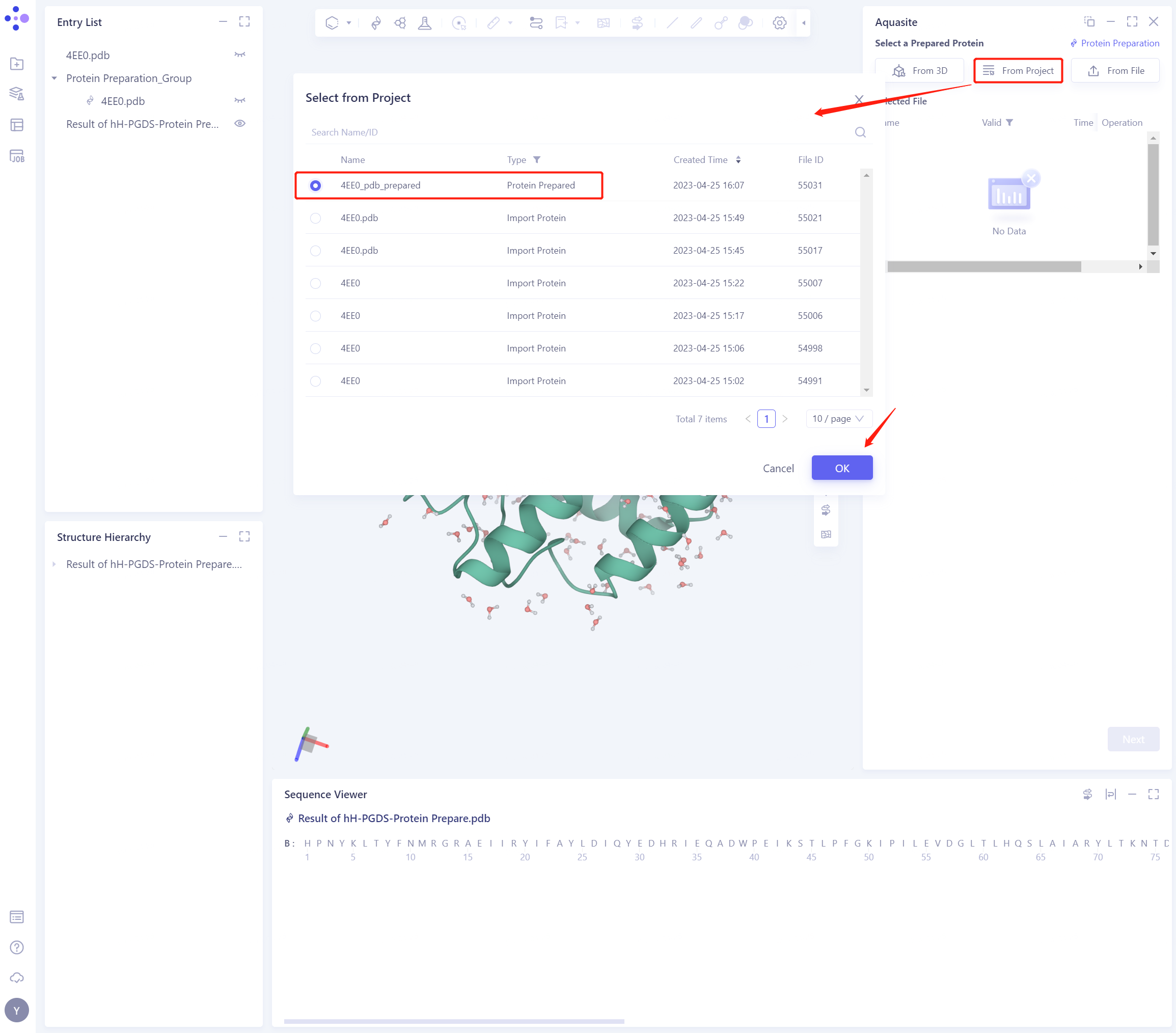
- After clicking OK, the system will automatically check whether the input protein meets the calculation requirements, and the status is “Processing”.
- In less than 1 minute, the system will judge that the protein is “Valid” and click “Next”.
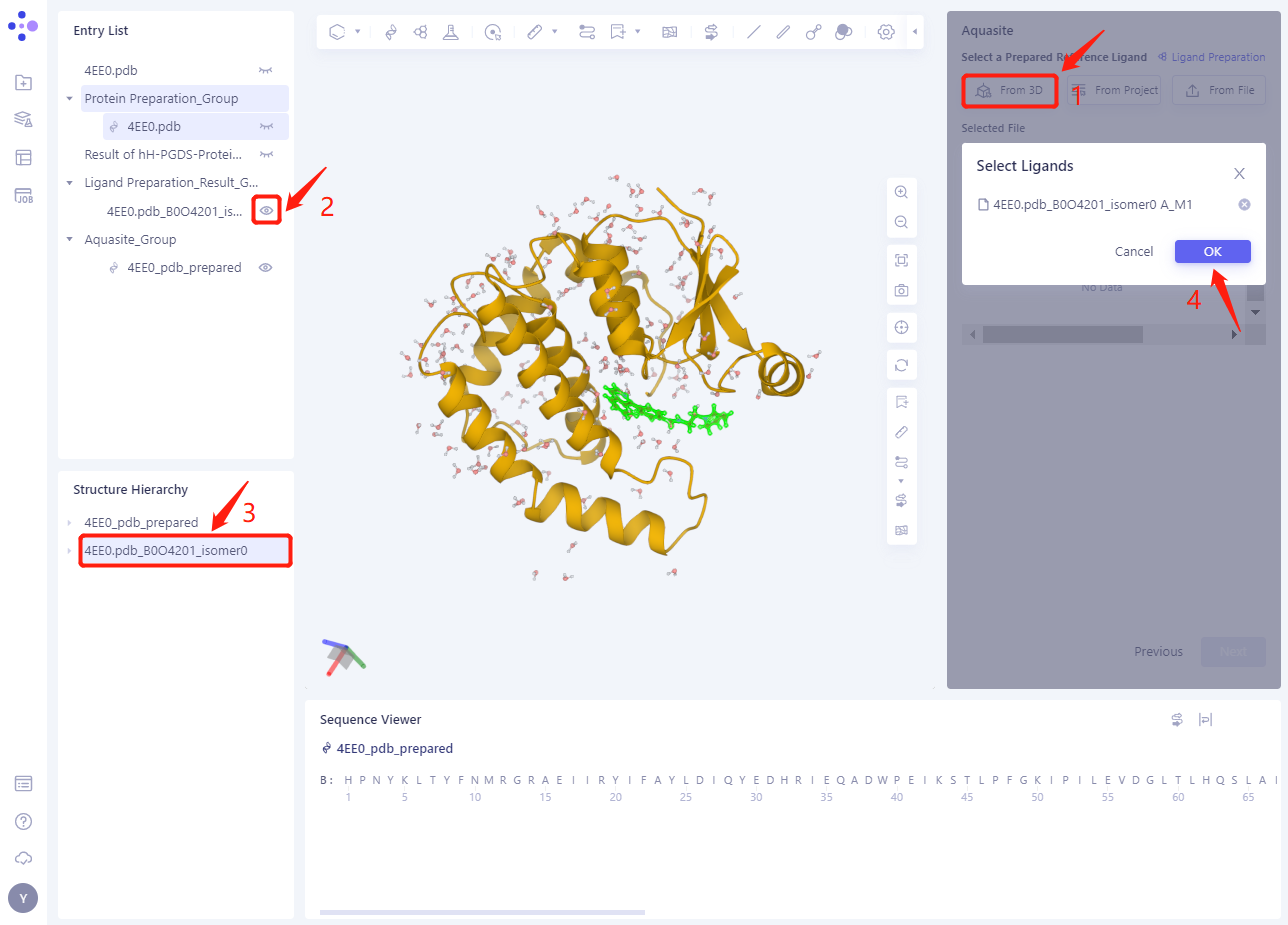
3.3 Select Prepared Reference Ligand
-
Click “From 3D”;
-
Click the structure show 4EE0.pdb _ B0O4201 _ isomer0 under the Ligand Preparation _ Result directory in the Entry List;
-
Select 4EE0.pdb _ B0O4201 _ isomer0 in the Structure Hierarchy ”.
-
Click OK in the Select Ligands “window.
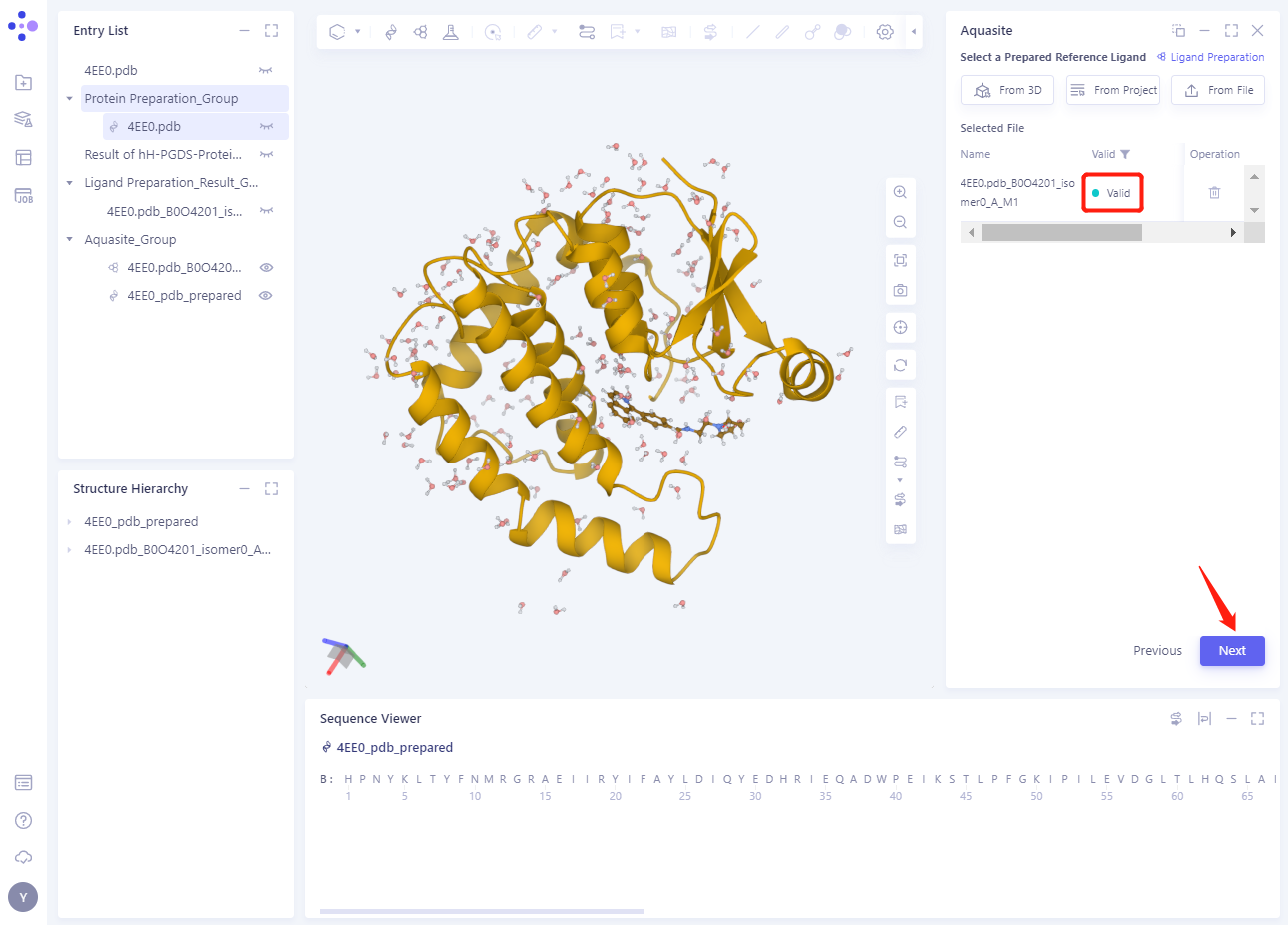
-
The ligand structure will be loaded into the Selected File ”. The system automatically checks whether the Ligand entered meets the calculation requirements and the status is “Processing”, and then the system judges that the Ligand is “Valid”.
-
Click Next
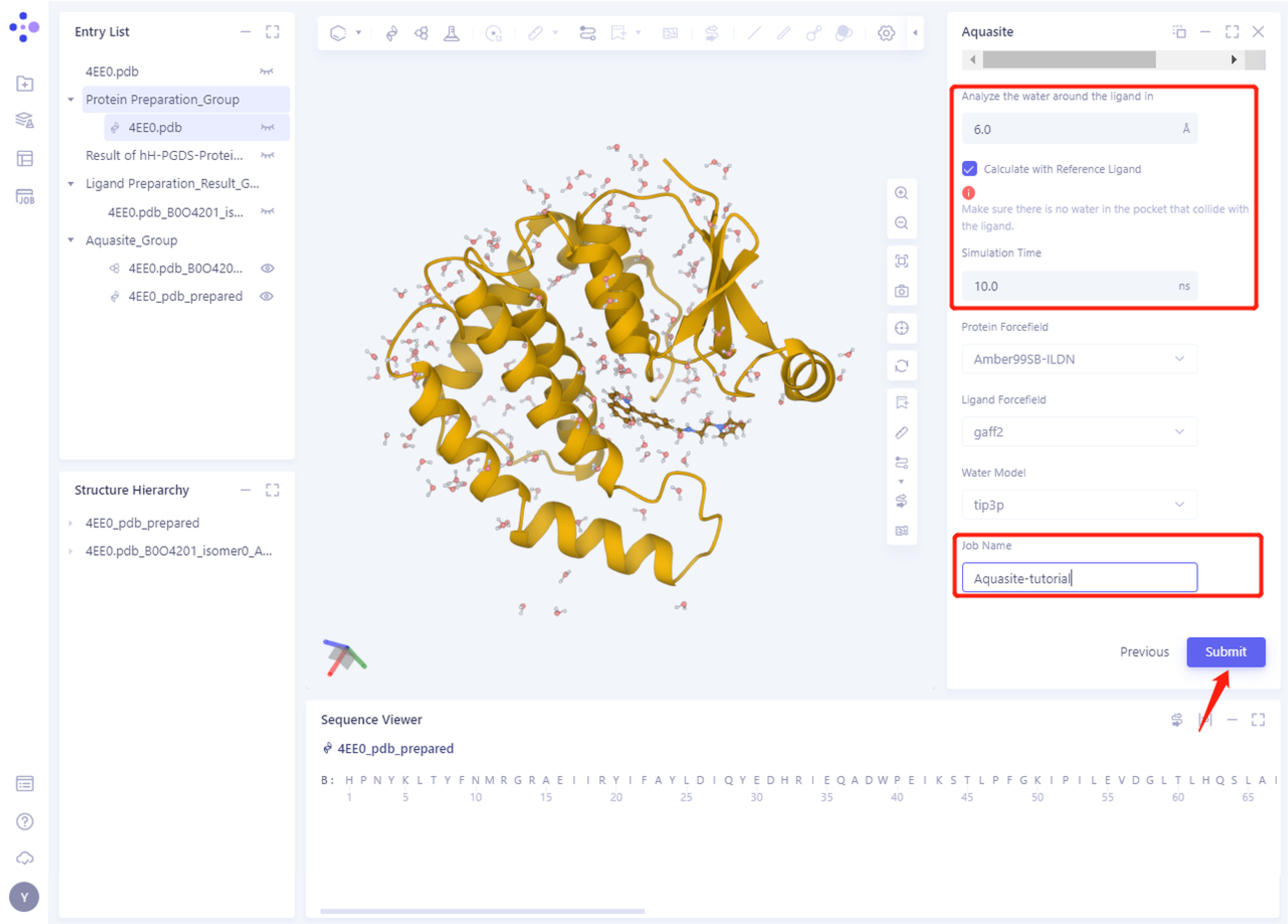
3.4 Confirm and Setting
-
The Analyze the Wate around the Ligand in “was modified to 6.0 (to ensure that the water molecules to be analyzed are within the specified range around the ligand);
-
Check the “Calculate with Reference Ligand” “option;
-
“Simulation Time” is modified to 10 ns (the longer the simulation time, the higher the calculation accuracy, and the calculation time will increase accordingly);
-
“Job Name” is named “Aquasite-tutorial”;
-
Leave the rest of the parameters at their default values and click “Submmit” to submit the task.
4. Analysis of results
4.1 Entrance
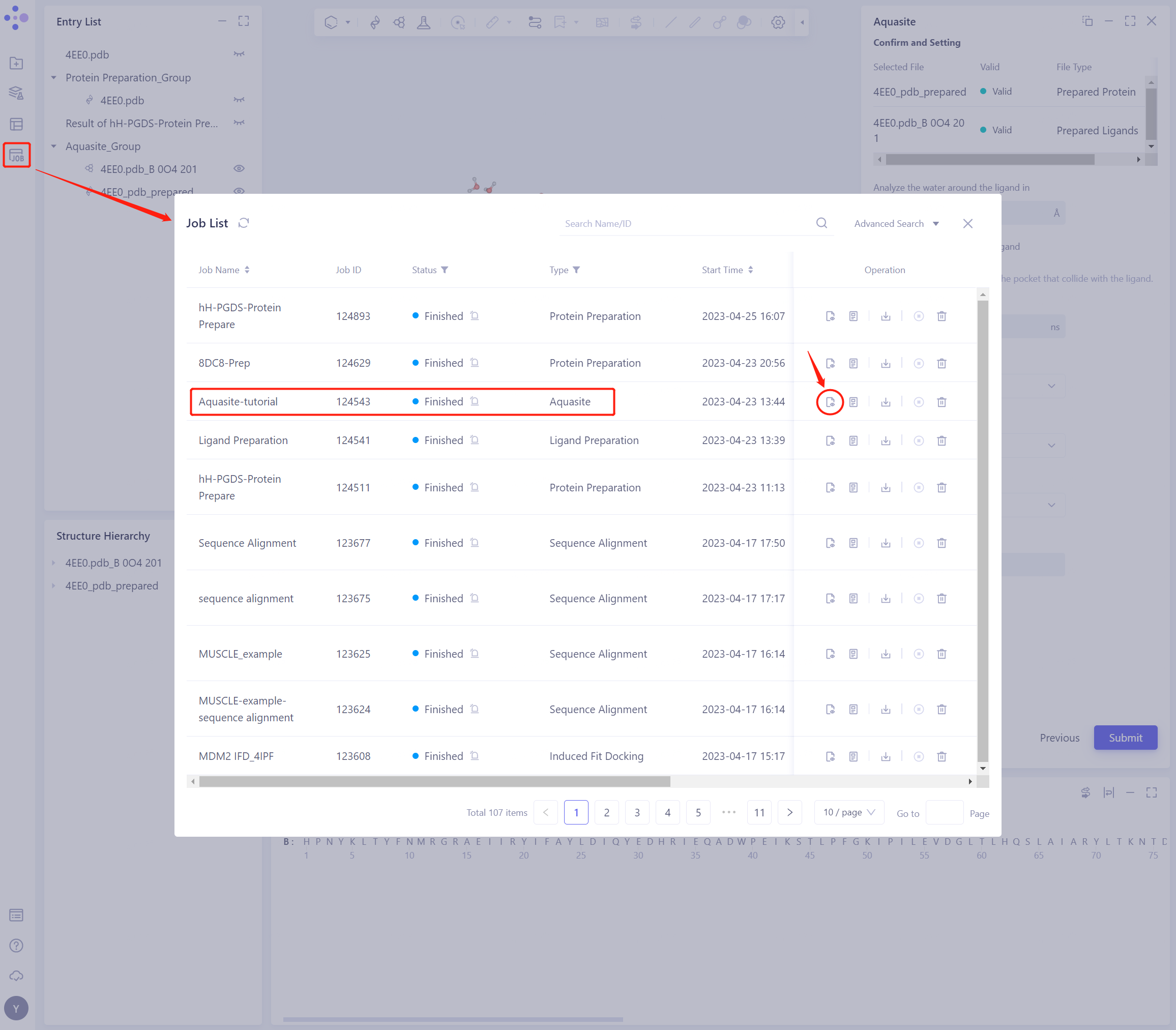
- In the left general menu bar Job → Job List, find the “Aquasite-tutorial” calculation task, and click “Show”
4.2 Results presentation
-
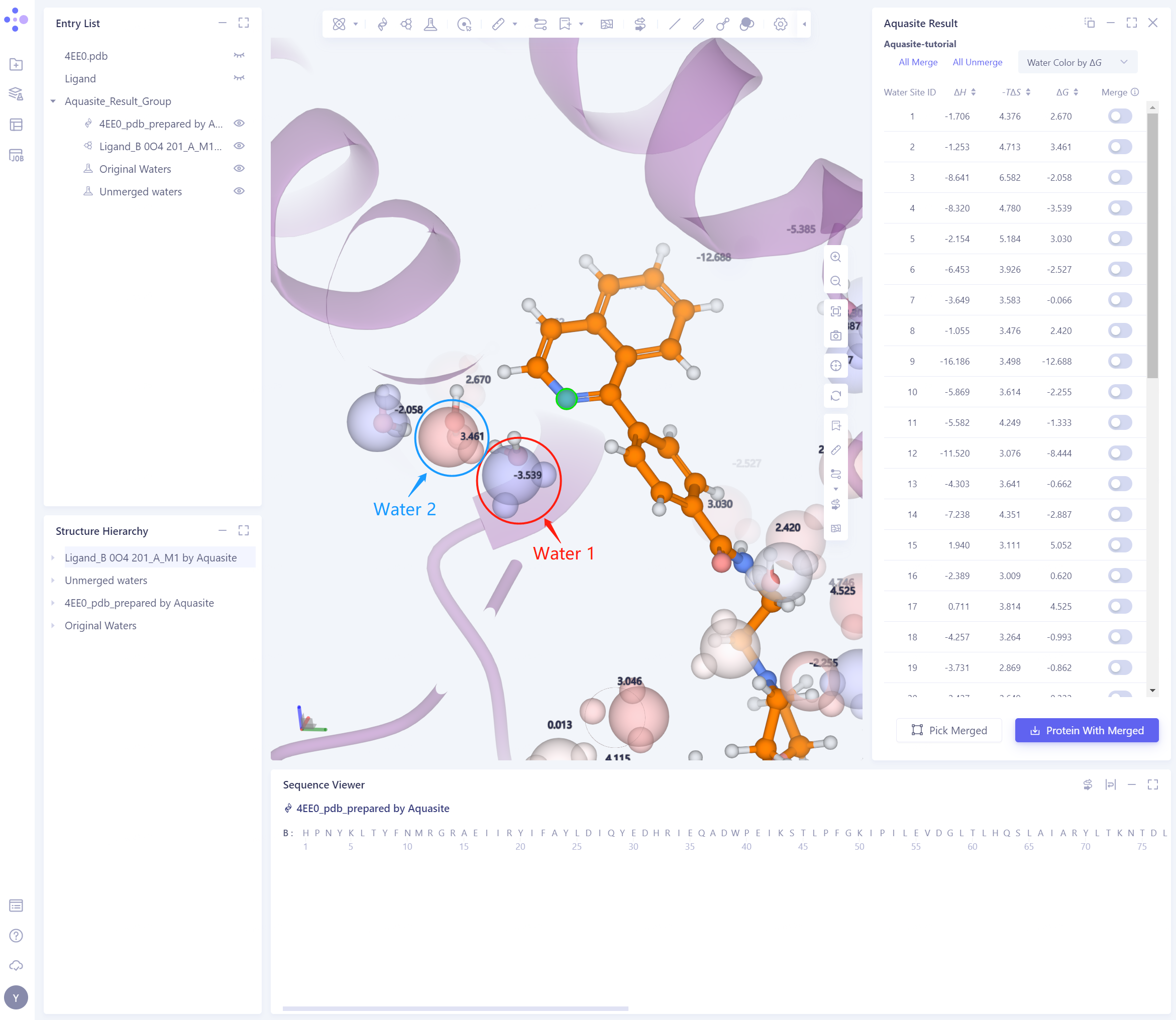
Adjust the molecular perspective of 3D Workspace to focus on the position and free energy of the ligand and predicted water water 1 (molecule in red circle), water 2 (molecule in blue circle) (co-crystal water molecules are shown in traditional Ball & Stick mode).
-
It can be seen from the results in the figure that the positions of the predicted water 1 and 2 are almost consistent with the positions of the eutectic water molecules in the respective marked circles. This indicates that Aquasite is relatively accurate in predicting the location of bound water.
-
The ΔG of water 1 is -3.539 kcal/mol and that of water 2 is 3.461 kcal/mol. This indicates that water 1 is more stable and important than water 2, and the substitution or replacement of this water molecule may have a greater impact on the binding affinity of the compound. In the experimental results of Trujillo et al., the change of R group will lead to a greater difference in activity, and our predicted results are consistent with the literature.
5. References
[1] Trujillo, John I., et al. “Investigation of the binding pocket of human hematopoietic prostaglandin (PG) D2 synthase (hH-PGDS): A tale of two waters.” Bioorganic & medicinal chemistry letters 22.11 (2012): 3795-3799.