使用教程(Tutorial)
1. 基本介绍
1.1 什么是自由能微扰(FEP)计算
自由能微扰(Free Energy Perturbation,FEP)计算是一种基于统计力学与分子动力学模拟的定量方法,用于评估分子体系在不同热力学状态之间的自由能差。其核心思想是通过构建热力学循环,将难以直接计算的结合自由能分解为可控的多步“变形路径”,从而精确预测小分子药物与生物大分子的结合亲和力。通俗地说,它帮助我们预测分子与目标蛋白结合时的结合强度,进而协助判断哪种分子可能更适合作为候选药物。FEP 广泛应用于药物设计中的先导物优化过程,尤其是在通过片段替换等方式改良先导化合物与蛋白的结合强度时,它能提供一种基于物理模拟的定量评估方法。
1.1.1 什么是相对自由能(RBFE)微扰计算
相对自由能(Relative Binding Free Energy, RBFE)微扰计算基于热力学循环原理,通过模�拟两个分子(如 A 和 B)在溶液中和目标蛋白结合状态下的自由能变化,来精确预测它们的相对结合自由能差异 ()。如图所示,FEP 的热力学循环分为两个部分:溶液环境和结合环境。
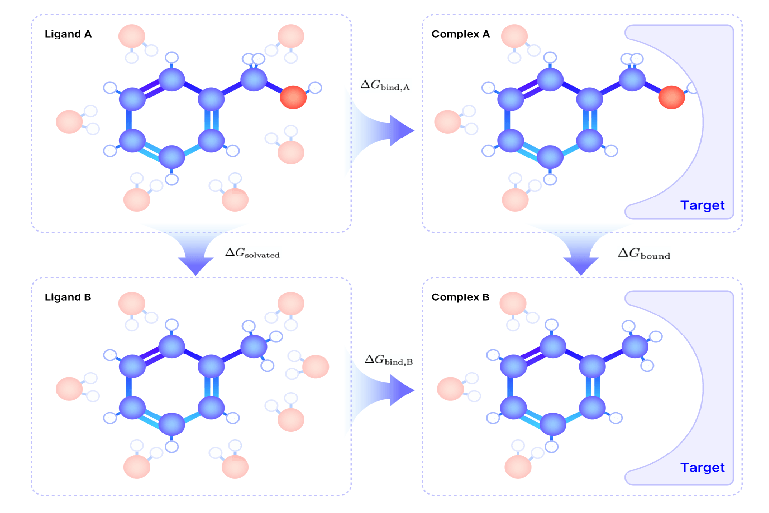
图:计算不同分子间相对结合自由能差的热力学循环
在溶液环境中,分子 A 和 B 与溶剂分子相互作用,分别计算其溶解自由能 ();在结合环境中,分子 A 和 B 与目标蛋白结合,分别计算其结合自由能 ()。然而,直接计算分子 A 和 B 的绝对结合自由能 (,) 是非常困难的,因为涉及溶剂化效应、分子间相互作用等复杂因素,容易受到误差累积的影响。
热力学循环为这一问题提供了解决方案——通过计算相对自由能差 (),即分子 A 和 B 在溶液和结合状态下的自由能差异,我们可以避免绝对自由能的计算,同时获得更加稳定和可靠的结果。根据热力学循环原理,两条路径(溶液和结合环境)上的自由能变化总是相等:
要实现上述热力学循环中的自由能差计算,RBFE 方法应运而生。RBFE 的基本思想是,通过分子动力学 (Molecular Dynamics, MD) 模拟,在分子 A 和 B 构建一个虚拟的“变形路径”,逐步将分子 A 的形状、性质或化学特性转换为分子 B。在这个过程中,记录下每一步中体系的自由能变化,最终通过统计力学公式将这些变化整合,计算出分子 A 和 B 的相对自由能差。通过以上步骤,RBFE 方法能够高效、可靠地完成复杂体系的相对自由能计算。
1.1.2 什么是绝对自由能(ABFE)微扰计算
当待测配体之间缺乏连续的化学空间、结构改动较大或根本不存在参考配体时,传统的相对自由能(RBFE)方法难以构建合理的逐步“变形路径”,也无法直接量化单一分子的结合亲和力。此时,绝对结合自由能(Absolute Binding Free Energy, ABFE)微扰计算便成为首选——它无需任何参考化合物,直接测定分子与目标蛋白结合时的结合自由能 ()。
然而,直接从分子在溶液态到蛋白结合态一次性计算十分困难:配体-蛋白和配体-溶剂之间的相互作用同时耦合,体系构象空间庞大,多种能量项的累积误差极易导致收敛性差、统计不稳;而且要在结合与非结合两种截然不同的环境中都达到充分采样,计算开销也非常高。
绝对自由能微扰计算(Absolute Binding Free Energy, ABFE)的核心思想,就是为了解决这一“难以一次性去耦合所有相互作用”问题,设计一个分步的热力学循环,将 的直接计算分解为复合物环境和纯溶液环境下的多步“去耦合/重耦合”过程。通过逐步移除或恢复配体与周围环境的相互作用,并在必要时施加或撤销空间与方向约束,既能保证各步之间构象重叠充分,又能有效降低一次性大规模去耦合带来的采样难题。
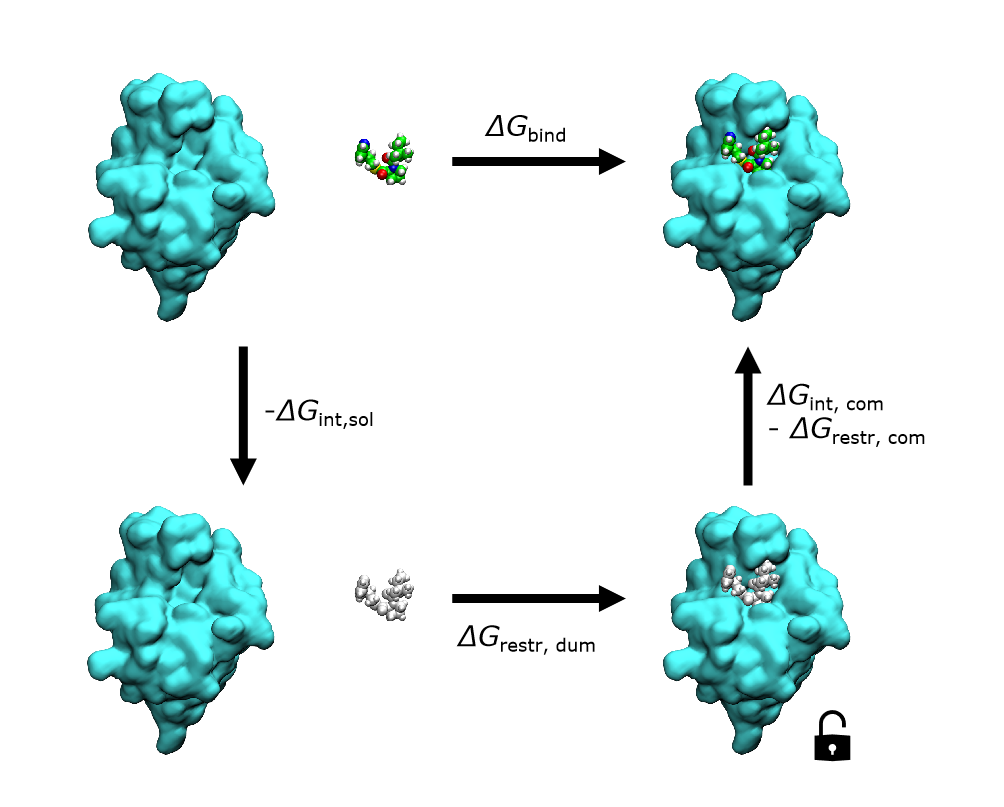
图:计算分子绝对结合自由能的热力学循环
如图所示,ABFE 热力学循环分为两个方向:(1)在蛋白–配体复合物环境中,配体与蛋白和溶剂的相互作用被分阶段去耦合,所得自由能变化记为 ;与此同时,为了限制配体在去耦合过程中的无序游离,需要解除先前施加的空间/方向约束,其对应的自由能变化为 ;(2)在纯溶液环境中,配体首先施加“虚拟约束”(dummy restraint),引入 ,随后逐步去耦合配体与溶剂的相互作用,得到 ;最后撤销虚拟约束,将配体释放至自由溶液态。
根据热力学循环原理,绝对结合自由能可表达为两条路径之差:
在实际计算中,需在足够密集的 λ 窗口下对上述每一步进行分子动力学采样,并利用 BAR、MBAR 等统计力学方法累积自由能增量;同时对位置/方向约束的能量贡献进行严格校正。虽然 ABFE 的计算成本和收敛要求高于 RBFE,但它无需任何参考配体,就能直接量化单一化合物的绝对结合热力学,尤其适用于结构改动较大或缺乏参照化合物的新型分子筛选场景。
思考:什么时候应该考虑使用ABFE而不是RBFE? 在实际研发中,当待测分子与参考配体仅在取代基或小基团上存在有限结构差异时,应优先采用相对自由能(RBFE)计算——其计算效率高、收敛速度快且统计误差较小;仅在分子改动幅度过大、难以构建可靠参照对,或确需获得单一配体的绝对结合自由能()时,才考虑使用绝对自由能(ABFE)计算;但需权衡的是,ABFE 对采样和约束校正的要求更严格,计算量显著增加,且可能带来更高的不确定性。 |
1.2 FEP能为先导物优化研发带来什么改变
在传统的先导物优化流程中,药化学家通常在已确定的核心骨架上,通过局部官能团改造来提升结合亲和力或优化分子性质。但由于结构改动幅度有限,绝大多数计算方法难以高精度预测微小替换对结合强度的影响,导致研究者只能先合成所有设计分子,再依次进行生物活性检测。这种“先合成、后筛选”的模式不仅耗时、耗资,而且在有限预算下,药化学家往往只能凭经验挑选部分候选物进行实验,极易遗漏具有潜力的分子。
引入基于自由能微扰(FEP)的方法后,先导优化迎来质的飞跃:研究者可以在任何实验合成之前,先对设计分子进行高精度的结合自由能计算,仅将 FEP 预测位居前列的分子投入合成与实验,从而显著降低合成与测试分子的数量。与此同时,FEP 并行化计算能力允许在相同时间内评估更多候选物,显著扩大可探索的化学空间。实践表明,在评估相同数量分子的项目中,FEP 的应用可将实验成本压缩约 76%、缩短实验周期约 79%;在预算不变的前提下,其并行筛选优势可使化学空间探索范围提升近 11 倍。
FEP计算正在将先导物优化实践从“经验驱动”带入“计算驱动”的新模式。
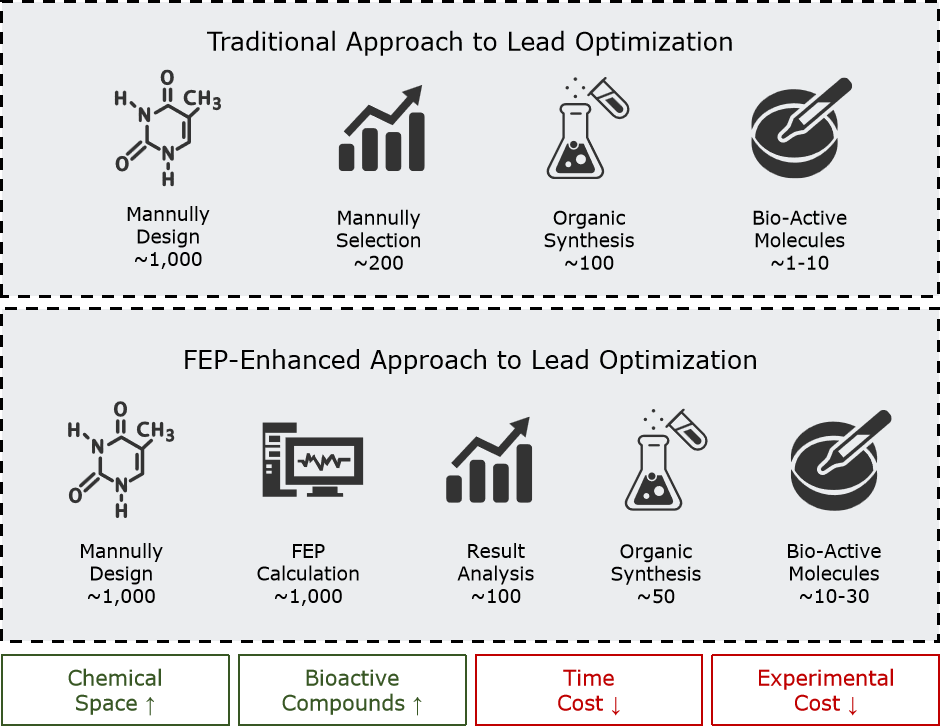
图:FEP计算带来的先导物优化方式改变
| 思考:为什么说FEP是先导化合物优化的利器? FEP 计算以接近化学精度(误差通常小于 1 kcal/mol)的预测能力,成为先导化合物优化中的核心工具。与传统依赖化学合成和实验筛选的方式相比,FEP 在虚拟环境中运行,可以快速评估大量候选分子,从而大幅增加探索的化学空间,降低优化过程中的时间成本。通过计算筛选代替实验验证,研究者可以减少大量化合物的合成和测试工作,将资源集中在最有潜力的分子上,显著提高发现活性化合物的机会。 |
1.3 什么样的项目适合使用FEP
FEP 的核心优势在于能够通过分子动力学模拟,以接近化学精度的水平预测分子与蛋白的结合自由能差异 ()。这一方法的有效性依赖于多个因素,包括目标体系是否具备清晰的结合模式、分子间的化学差异是否在可控范围内,以及实验数据的质量是否足够支撑计算模型。因此,在使用 FEP 之前,需要确认以下条件是否满足:
目标体系具有高质量的蛋白-小分子共晶结构
共晶结构分辨率优于 2.2 Å:体系中的每个氨基酸残基和配体小分子应具有明确的几何位置,结合位点的结构完整且细节清晰。
配体小分子的结合模式明确:配体小分子需与蛋白形成合理的相互作用(如氢键、疏水作用等),并符合已知的结合机制;结合位点内的关键氨基酸残基侧链在结构中应无缺失且位置明确。
| 思考:如何应对缺乏高质量共晶结构的情况? 当体系缺乏高分辨率的共晶结构时,可以结合多种建模手段,如使用 AlphaFold 预测蛋白结构、通过分子动力学模拟优化结合构象,或者采用分子对接和诱导契合对接方法生成初始模型。然而,所有生成的结构都需要经过严格验证,特别是配体在结合位点中的结合模式是否合理,是否符合已知的相互作用机制。如果共晶结构中某些区域未解析且远离结合位点,可通过计算工具补全;对于额外的结构区域,如果与配体结合无关,也可以适当简化。遇到复杂的建模需求时,建议咨询专业的计算生物学团队,以确保初始模型的可靠性和后续计算的准确性。 |
待评估的配体集合小分子间结构相似
配体小分子化学差异的改动范围有限:配体小分子集合需在结构上形成连�续的化学空间,每个分子至少与集合中的其他分子有显著相似性;允许的化学改动包括取代基的变化、骨架跃迁、开关环或环大小的调整,但整体形状不能发生显著变化。
配体小分子与蛋白的结合模式一致:所有配体小分子在结合位点中应形成一致的关键相互作用网络(如共享的氢键或 π-π 堆积);共享的关键子结构应在结合构象中保持叠合。
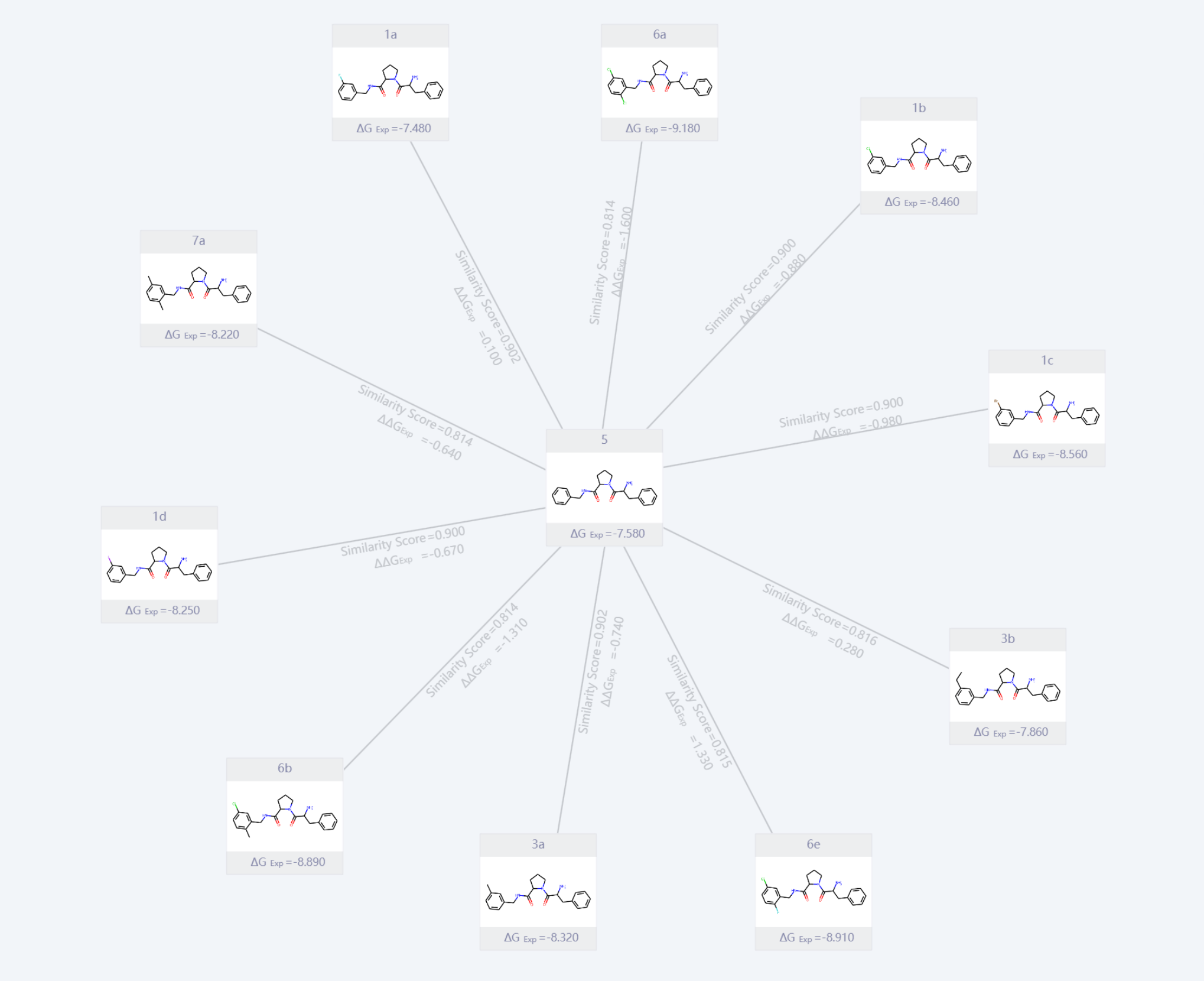
图:配体集合小分子间结构相似、化学空间连续
部分配体具有可靠的实验数据
实验数据的来源统一:实验数据需在一致的实验条件和批次下获得,包括相同的实验方法(如 SPR 或 ITC)、蛋白来源和缓冲体系。
实验数据覆盖较大结合亲和力范围:实验数据应涵盖从弱结合到强结合的范围,例如 IC50 值从毫摩尔到纳摩尔不等。
| 思考:为什么需要部分配体具有可靠的实验数据? 实验活性数据在 FEP 计算中扮演着验证和校准的双重角色。正如实验需要标准对照一样,计算方法在应用于实际预测前,必须首先通过实验数据确认其在目标体系中的适用性。通过对一组具有已知结合亲和力的分子进行计算和对比,能够验证 FEP 的预测精度,并调整参数以确保其结果与实验数据一致。只有在确认计算方法能够准确捕捉体系的结合特性后,FEP 才能被用于筛选和优化新的化合物。这一过程确保了计算的可靠性,同时提升了对未测定分子的预测能力。 |
总的来说,FEP 是一种强大的计算工具,但其成功应用依赖于体系是否满足关键条件。如果体系具备高质量的结构数据、配体集合的化学特性合理、实验数据支持充分,FEP 将能够高效地为先导化合物优化和分子设计提供可靠的预测能力。
在实际项目中,如果您不确定体系是否满足 FEP 的适用条件,可以结合本文的指南逐一评估,并咨询团队内的专家或参考已有的文献模型。如果条件初步符合,建议从小规模的测试体系开始尝试,通过基准测试验证预测精度,再逐步扩展应用范围。这样的实践方法可以确保资源的有效利用,同时降低模型构建过程中的风险。
| 思考:什么样的项目一定不适用FEP? 蛋白-配体分子结合模式尚不确定 配体集合的化学差异过于剧烈 目标蛋白的动态特性过于复杂 |
1.4 将FEP方法应用于项目的一般步骤
将 FEP 方法应用于药物设计项目时,通常分为两个阶段:验证阶段和生产阶段。这种分阶段流程旨在确保模型的可靠性,同时提升资源利用效率。在验证阶段,研究的重点是测试已知配体的预测精度,确认 FEP 方法是否适用于目标体系,并构建可用的 FEP 计算模型;在生产阶段,则利用验证后的模型预测未知分子的结合能力,为分子筛选和优化提供支持。
| 验证阶段 回顾性研究(Retrospective study) | 生产阶段 前瞻性研究(Prospective study) | |
| 输入 | 已测定实验活性的配体分子集合 | 药化学家设计的待测定活性的候选配体分子集合 |
| 任务 | 验证FEP计算模型精度、优化蛋白-配体复合物结构和模拟参数 | 利用验证通过的 FEP 计算模型预测候选分子的结合��自由能 |
| 输出 | 验证通过的 FEP 计算模型 | 供实验验证的优选分子列表 |
验证阶段:研究人员需要选择一组具有实验活性数据的已知配体,并确保这些数据覆盖弱、中、强结合的亲和力范围。这一阶段的核心目标是评估 FEP 方法在目标体系中的适用性,确保其预测结果具有足够的精度和可靠性,同时通过调整参数设置(如力场、模拟条件和结合模式)来优化模型。具体而言,验证过程首先通过 FEP 模拟计算已知分子的相对结合自由能差 (),将计算结果与实验数据进行对比,使用统计方法(如 RMSE 和相关系数 )评估模型的精度。
如下图所示,横轴表示实验测定的结合自由能 (),纵轴表示 FEP 计算的结合自由能 ()。图中的回归线表示计算值与实验值之间的线性拟合程度,而数据点的误差棒则反映了每个分子的计算不确定性。深蓝色区域代表较高置信区间(± 1 kcal/mol),表明计算结果与实验数据高度一致的范围,而浅蓝色区域则涵盖了更广的不确定性范围(± 2 kcal/mol)。一般来说,更多数据点落在深蓝色区域内,表明模型预测误差较低,整体精度较高。若多数数据点偏离深蓝色区域,可能存在模型误差或参数问题,需要进一步优化。
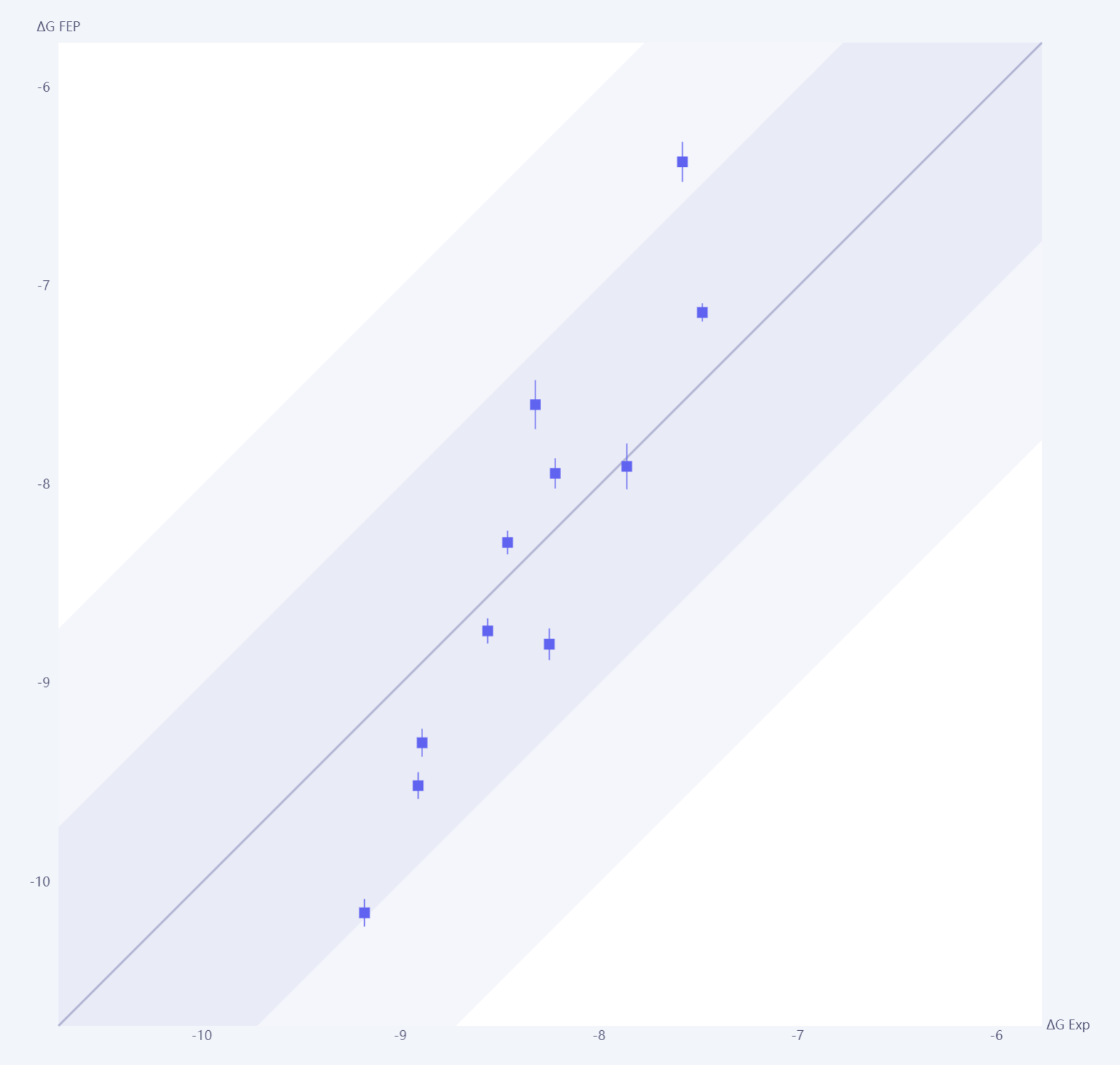
图:FEP计算得到的自由能值和实验测定活性值的相关性图
通过迭代优化力场参数、结合模式和模拟条件,确保计算结果与实验数据达到一致性。验证完成后,构建好的 FEP 模型可进入生产阶段,支持后续的分子筛选和设计工作。
| 思考:什么是 FEP 计算模型? FEP 计算模型是指用于自由能微扰计算的完整体系,包含构建 FEP 方法所需的关键组件。具体而言,这包括以下几部分: 蛋白-配体复合物结构:经过预处理的目标蛋白结构、配体分子结合姿态,为后续自由能计算提供准确的初始状态。 模拟参数:用于描述体系的蛋白、配体分子力学力场,以及模拟条件(如温度、溶剂模型和路径构建方法)。 |
生产阶段:通过验证后的 FEP 模型,可以用于开展大规模分子筛选或优化工作。此时的输入分子集合通常由药物化学家设计,从苗头化合物出发设计的一系列结构衍生物。通过 FEP 计算,对候选分子逐一计算其相对结合自由能 (ΔΔG),筛选出结合自由能较低、潜在活性更高的分子。这些优选分子将用于进一步实验验证,同时计算结果为分子设计迭代提供反馈。生产阶段依托验证阶段的高精度模型,实现高效可靠的分子筛选,显著缩小实验筛选范围并提升药物研发效率。
1.5 关于Hermite®️ Uni-FEP
Hermite®是深势科技打造的AI for Science加持的新一代药物计算设计平台,为临床前药物研发提供一站式计算解决方案,包含行业领先的自由能微扰计算工具Uni-FEP、超高通量虚拟筛选工具Uni-VSW等核心模块,赋能蛋白结构预测、靶标确证、苗头化合物发现、先导物优化等多个环节。Hermite®提供了基于网页的交互式全新分子展示体验,能对项目、成员、数据进行细粒度管理,具有完备资质认证和多层级安全防护,支持在线使用和私有化部署。Hermite®平台深受客户信赖,在中国已有超六成行业头部企业选择使用Hermite®平台,应用于超过50个药物管线项目中。
| 小贴士:使用Hermite®️药物计算设计平台的硬件、软件建议 硬件:推荐使用23.5 英寸及以上的显示器,以便更清晰地展示蛋白-配体复合物结构、结合模式、配体叠合状态,以及微扰图和原子映射图等信息,提升建模效率和分析精度;建议计算机配置 8GB 及以上内存,以确保软件运行流畅、操作无卡顿。 软件:建议使用最新版本的 Google Chrome 浏览器,以确保平台的各类显示组件正确加载并顺畅运行。 |
Uni-FEP是深势科技开发的一��款基于自由能微扰(Free Energy Perturbation,FEP)理论的计算辅助药物设计工具,旨在精确评估蛋白质与配体之间的结合亲和力。Uni-FEP结合了分子动力学、增强采样算法和分子力场的AI优化技术,在超过50个蛋白体系上经过验证,达到了行业领先水平。经过高性能计算优化,Uni-FEP在推荐参数下的单次FEP计算可在约4小时内完成;依托海量云算力,Uni-FEP可以同时开展数千个FEP任务的并行计算。同时,Uni-FEP提供基于浏览器的直观可视化交互界面,能可视化地检查和调整分子结构、蛋白-配体复合物结合模式,自由地构建和编辑微扰计算图和原子映射,并多维度地分析FEP计算结果,提供了业内领先的交互体验。商业客户已累计开展Hermite® Uni-FEP计算任务超过20万余次。
| 小贴士:Hermite®️ Uni-FEP模块计算精度验证案例 第六章节详细介绍了Hermite®️ Uni-FEP模块用于验证和展示结合自由能计算精度的案例,感兴趣的读者可以到第六章详细了解。其中,在验证体系[1](共计8个蛋白-配体分子集、199个分子)上,Uni-FEP经过5ns模拟计算得到结合自由能与实验活性的相关性系数R2均值为0.56,均方根误差为0.89 kcal/mol;在验证体系[2]上(共计8个蛋白-配体分子集、264个分子),Uni-FEP经过5ns模拟计算得到结合自由能与实验活性的相关性系数R2均值为0.41,均方根误差为1.23 kcal/mol。这些测试表明Uni-FEP计算精度达到业内领先水平。
|
Hermite®️ Uni-FEP模块是专门面向自由能微扰计算场景设计的药物计算设计工具,由FEP任务提交、FEP任务管理和FEP结果分析三个主要页面组成,提供了复合物三维结构窗格、微扰计算图平面窗格、原子映射二维校验和三维修改窗格等交互方式,支持一站式的体系准备、微扰计算图构建、原子映射检查、模拟参数设置、运算任务管理、模拟结果查看、结合自由能统计分析等完成FEP计算任务的全部功能。
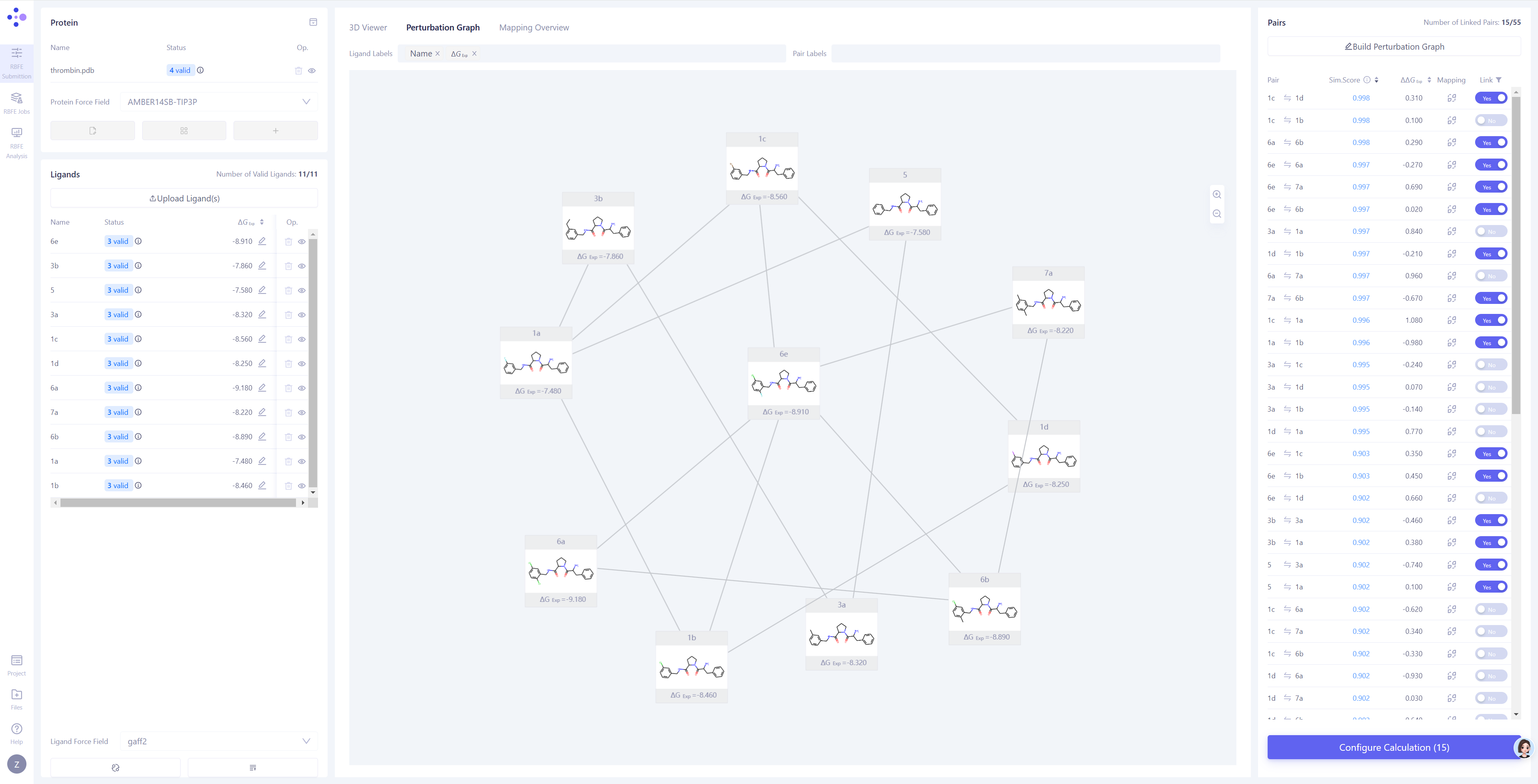
图:Hermite®️ Uni-FEP模块任务提交界面
2. 使用Uni-FEP进行RBFE计算 - 操作清单
| 小贴士:不熟悉RBFE计算的读者可以先跳过本章 本章节旨在给熟悉RBFE计算的用户一个清晰简要的使用指南,以确认每一个重要步骤都完成操作。 如果您首次接触RBFE计算,可以先阅读第三章节,通过案例和可视化界面截图了解如何使用Hermite Uni-FEP完成一次完整的RBFE计算和分析工作。再以本章节的内容作为实操过程的指引。 |
2.1 RBFE 数据收集与基本检查
收集并检�查蛋白结构
选择高质量的晶体复合物结构作为起始蛋白质模型,确保结合口袋附近的氨基酸残基清晰解析,蛋白质链无断裂。
确认参考配体与结合口袋形成合理的相互作用模式。
收集并检查分子结构和性质
确保分子集中的化合物与参考配体共享部分子结构,且在结合口袋中的结合模式与参考配体一致。
确保部分分子具备可靠的实验数据,且数据来源统一,覆盖较大的结合亲和力范围。
2.2 RBFE 体系准备与任务提交
上传蛋白并进行准备
上传蛋白结构并进行蛋白预处理流程,保留必要的氨基酸链、水分子和其他辅助结构,设定氨基酸的质子态。
蛋白预处理后,系统会进行自动检查。若检查未通过,根据反馈修正结构。
若结合位点中存在关键的可质子化氨基酸残基(如Asp、Glu、His等),请仔细确认并根据结合模式手动调整其质子化状态,以确保计算的准确性。
如蛋白系统中包含对功能或结合模式具有重要影响的辅因子(如NAD、FAD、金属离子等),请将其一并上传并确认其结构与状态的合理性。
若目标蛋白为膜蛋白,请在预处理后搭建合适的磷脂双分子层。
上传配体分子并进行分子对齐
上传配体分子并进行自动检查。若检查未通过,根据反馈结果修正结构。
对于未与参考分子叠合的配体,设定与参考分子的公共骨架以进行分子叠合。若叠合效果不佳或与蛋白质结构发生碰撞,需修改公共骨架或选择其他参考分子重新叠合,直至所有分子形成保守且合理的结合模式。
如配体分��子具备实验测定的结合亲和力数据,需导入该数据。
构建微扰计算图并检查待计算分子对的原子映射
基于配体分子信息,更新微扰分子对列表。
自动生成微扰计算图,或根据分子间的相似性手动构建,确保所有配体分子至少与一个结构相似的分子连接。
仔细检查每一对待计算分子的原子映射,确保公共结构的原子一一对应。
设定计算参数并提交计算任务
进入提交前的确认页面,核实所使用的蛋白质和待计算的分子对是否符合预期。
检查并确认 RBFE 计算的模拟参数,确保设置正确。
确认即将消耗的 FEP pairs 数,确保账户余额足以完成本次计算。
点击提交,RBFE 计算任务将自动分配至计算集群。
2.3 RBFE 任务监控与结果分析
监测任务进程和状态
在 RBFE 任务界面,监控每一对分子对的模拟进程和状态。
若某个分子对的模拟失败,需检查并修改其原子映射或模拟参数,考虑重新提交。
分析单个分子对计算结果
模拟任务完成后,展开具体结果,观察模拟是否收敛。
如未收敛,考虑重新提交或延长模拟时间。
若一个分子对有多次模拟记录,应根据收敛性选择最优结果作为后续分析的基础。
汇总所有分子对的计算结果
进入 RBFE 分析界面,根据热力学循环的质量判断应保留哪些分子对用于最终结果分析。
通过相关性图,查看 RBFE 计算所得的结合自由能与实验数据的相关性,评估 RBFE 计算模型的可信度。
基于 RBFE 计算结果,对未测定活性的分子进行计算评估。
3. 使用Uni-FEP进行RBFE计算 - 以Thrombin体系为例
Thrombin 蛋白是一种在血液凝固过程中起关键作用的酶。在凝血级联反应中,它通过将纤维蛋白原转化为纤维蛋白,从而形成血凝块。抑制或调节 Thrombin 的活性可以直接影响血液凝固过程。因此,开发 Thrombin 蛋白的小分子抑制剂对于治疗血栓类疾病和抗凝药物至关重要。
在本章节中,我们将基于已报导的药物研发项目,以Thrombin 蛋白为目标,展示如何利用 Uni-FEP 进行自由能微扰计算的体系构建与验证。本案例来自 Bernhard Baum 等人2009年于 Journal of Molecular Biology 发表的研究[1],该工作深入探讨了药物分子在 Thrombin S1 特异性口袋中的结合机制,结合 X-ray晶体结构解析 等多种实验手段,揭示了Asp189和Tyr228等关键残基形成的相互作用在结合自由能中的关键贡献。
| [1] Baum B, Mohamed M, Zayed M, Gerlach C, Heine A, Hangauer D, Klebe G. More than a simple lipophilic contact: a detailed thermodynamic analysis of nonbasic residues in the S1 pocket of thrombin. Journal of molecular biology. 2009 Jul 3;390(1):56-69. |
3.1 RBFE 数据收集与基本检查
本教程所使用示例数据可以在通过链接 https://bohrium-api.dp.tech/ds-dl/uni-fep-tutorial-j234-v1.zip 下载获得。
3.1.1 收集并检查蛋白结构
我们选取了 2ZFF 晶体结构作为参考模型,该结构解析了 Thrombin 蛋白与未取代苯基抑制剂 5 的复合物,分辨率高达 1.47 Å,提供了清晰且高质量的结合口袋构象,尤其是 S1 特异性口袋中关键残基(如 Asp189 和 Tyr228)的空间位置。这为 FEP 计算提供了可靠的起始蛋白结构,并确保结合口袋内的相互作用模式具有较高可信度。

图:Thrombin 蛋白与抑制剂的复合物晶体结构(PDB ID:2ZFF)
3.1.2 收集并检查分子结构和性质
我们选择了结构相似但取代基不同的一系列抑制剂,包括 1a, 1b, 1c, 1d, 3a, 3b, 5, 6a, 6b, 6e 和 7a。这些分子均保留了与蛋白结合高度一致的核心骨架,而在 S1 口袋内的苯基取代基表现出系统性的变化,例如氯、氟、溴和甲基等取代基,能够分别反映疏水性、体积及特殊相互作用对结合驱动力的贡献。此外,这些分子覆盖了实验测定的较宽结合自由能范围,为 FEP 模型的验证提供了完整且统一的热力学数据支持。
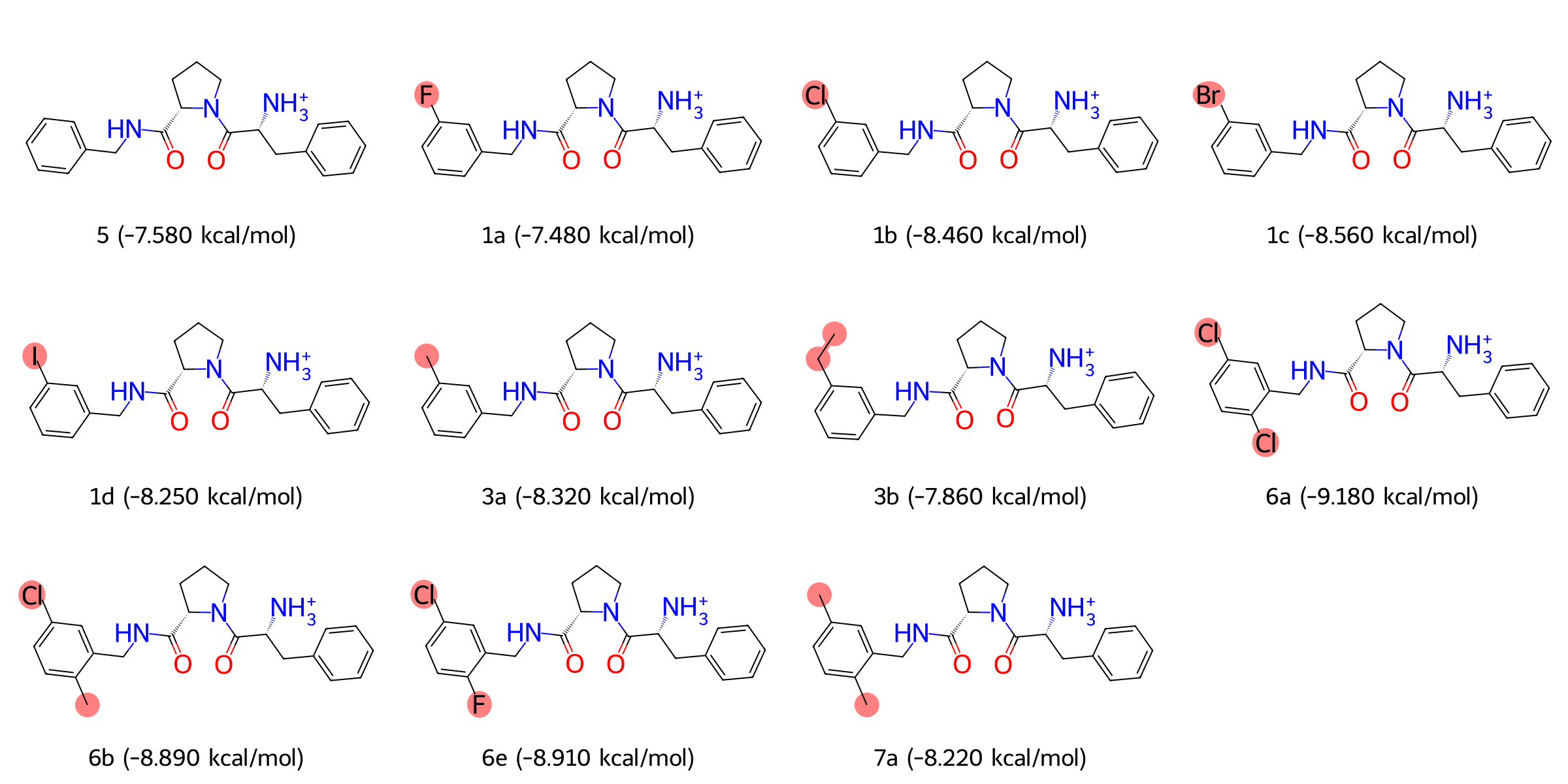
图:Thrombin 蛋白抑制剂的化合物结构和结合自由能
3.2 RBFE 体系准备与任务提交
3.2.1 创建FEP项目并进入项目页面
| 操作描述 | 界面截图 |
| 在 Hermite 项目列表 页面中,点击右上角的 "+ New" 按钮,输入项目名称(如 "Tutorial_Thrombin"),选择项目类型为 "FEP",然后点击 "Apply",即可创建一个 FEP 项目并进入项目页面。 |
|
3.2.2 上传蛋白并进行准备和检查
蛋白质准备是 FEP 计算的重要步骤,确保蛋白质结构的合法性、准确性和稳定性。本小节将分步骤指导您如何完成蛋白质的上传、结构检查、质子化调整等操作。
| 操作描述 | 界面截图 |
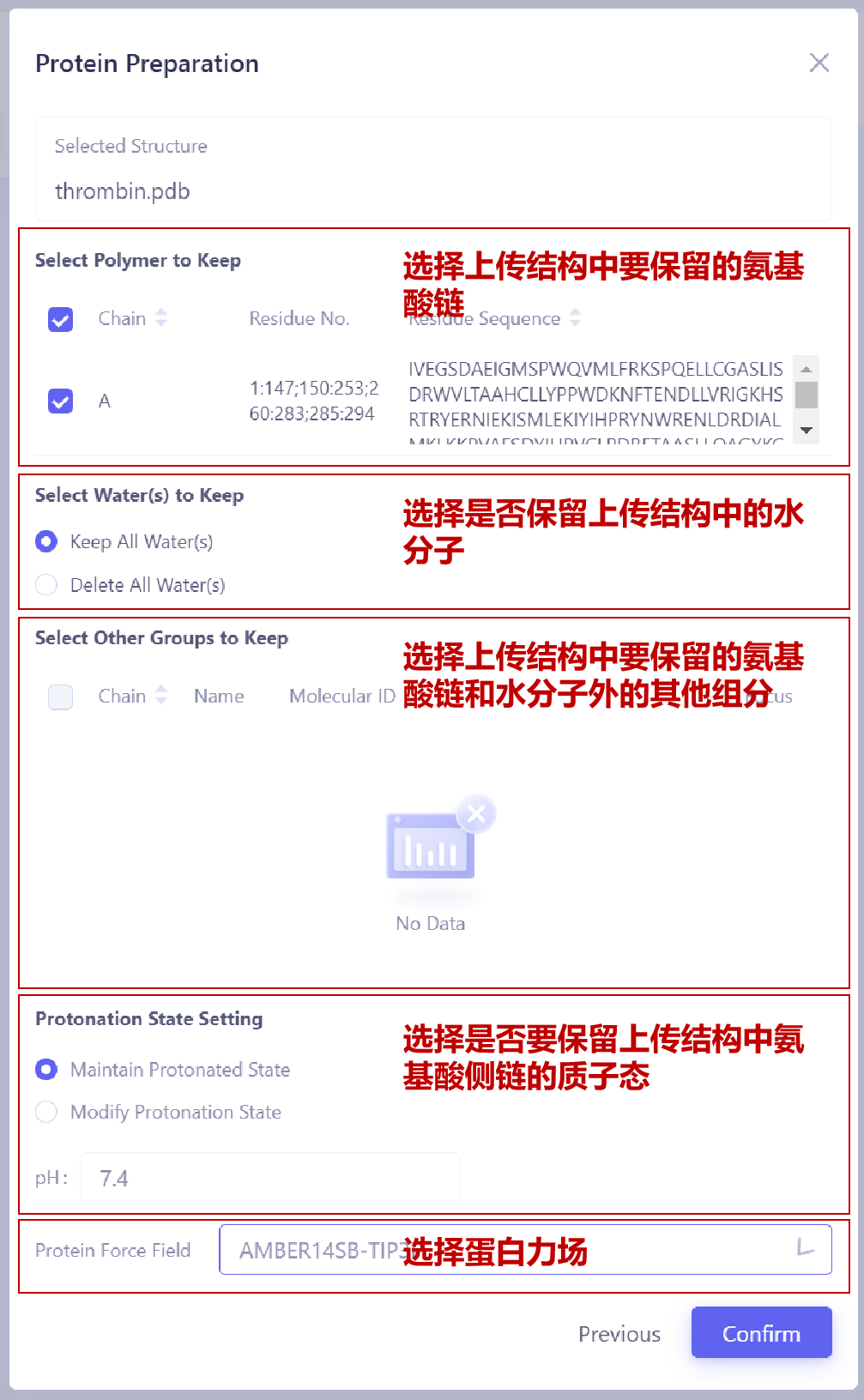
上传蛋白结构文件并进行准备:在页面左上角蛋白窗格,点击 Upload a Protein 按钮,选择要上传的蛋白结构文件(例如 thrombin.pdb),然后点击 Next 进入蛋白准备界面。选择保留的氨基酸链:在 Select Polymer to Keep 选项中,选择需要保留的氨基酸链。一般来说,应保留与配体结合位点相关的氨基酸链,以确保后续计算的准确性。 选择保留的水分子:在 Select Water(s) to Keep 选项中,选择是否保留晶体结构中的水分子。如果上传结构中存在:(1)深埋在蛋白内部且对整体结构稳定性重要的水分子; (2)参与蛋白-配体相互作用的水分子,则建议选择 Keep All Water(s),否则可以选择 Delete All Water(s) 删除水分子,以减少计算负担。 选择保留的其他分子或化学基团:在 Select Other Groups to Keep 选项中,检查是否有需要保留的其他分子或化学基团(例如辅助因子、共价修饰基团等)。 调整氨基酸侧链质子化状态:在 Protonation State Setting 中,选择质子化状态设置。如果您已经确认氨基酸的质子化情况,使用 Maintain Protonated State,以保留晶体结构中氨基酸侧链的质子化状态。否则,您可以使用 Modify Protonation State 让程序根据您设置的pH值自动调整氨基酸的质子化状态。 选择蛋白质力场:在 Protein Force Field 中,选择合适的力场(例如 "AMBER14SB-TIP3P"),以确保在分子动力学模拟中蛋白的能量计算准确可靠。 确认蛋白结构设置:完成上述设置后,点击 Confirm 按钮,系统将开始蛋白结构的预处理和合法性检查。通常,这一过程会持续约1分钟。 |
|
| 检查蛋白准备状态:蛋白准备完成后,系统将显示检查结果状态,包括 "Valid"(有效)、"Warning"(警告)或 "Error"(错误)。您可以根据提示进行修正或进一步检查。 |
|
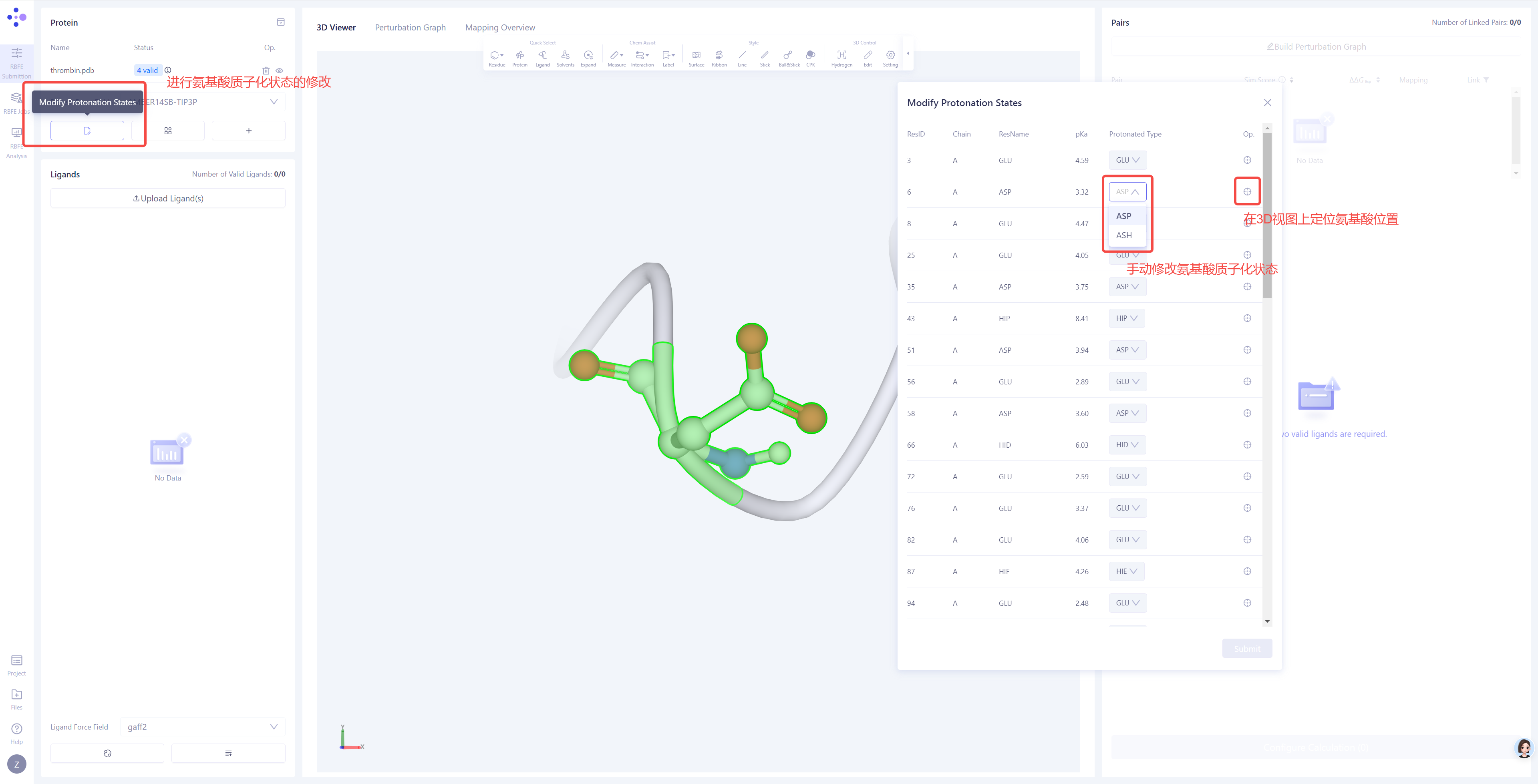
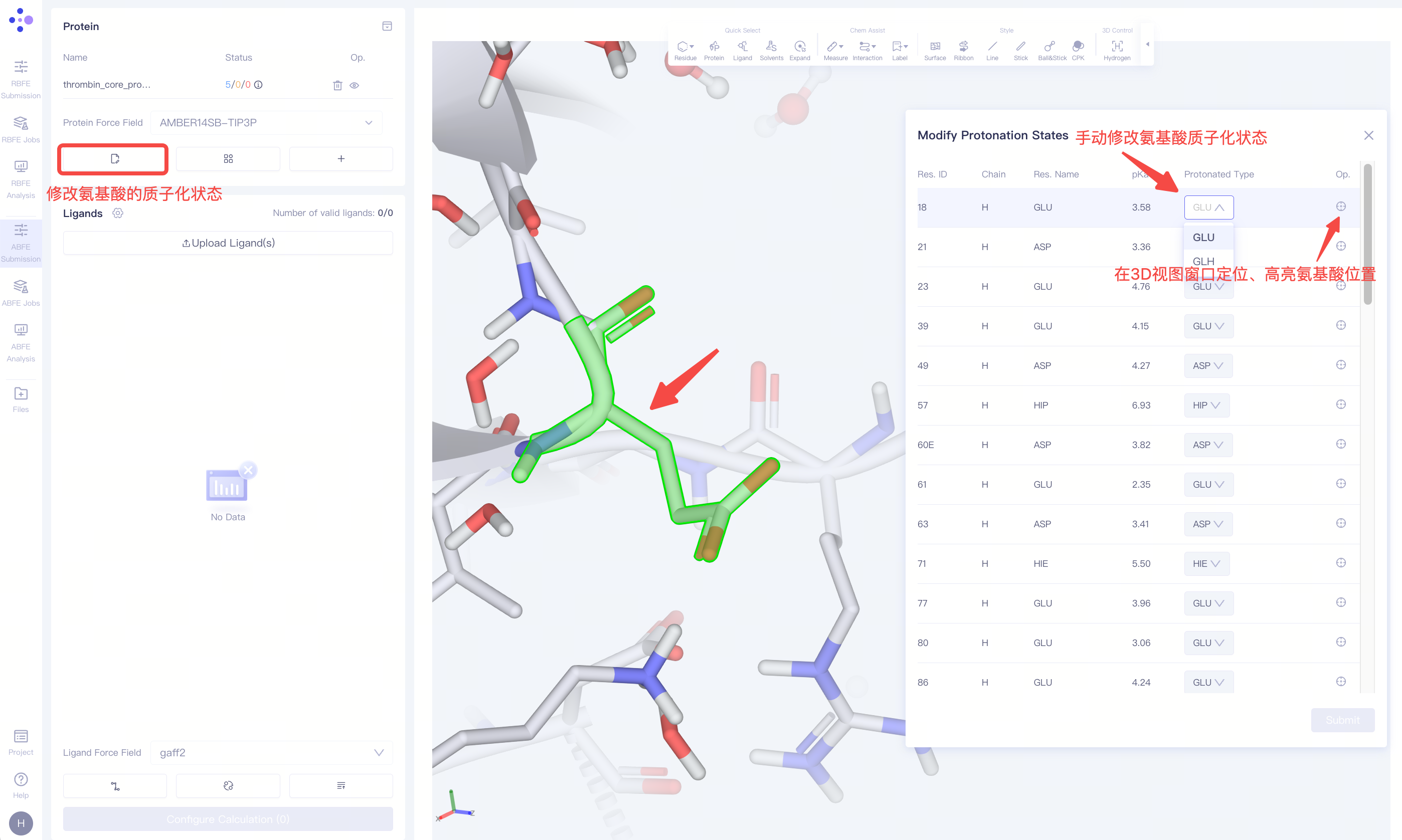
手动调整质子化状态:如果检查结果中存在质子化状态的警告或蛋白配体结合位点附近有重要的可质子化氨基酸,您可以点击 Modify Protonation States 选项,对质子化状态进行手动检查和调整。 |
|
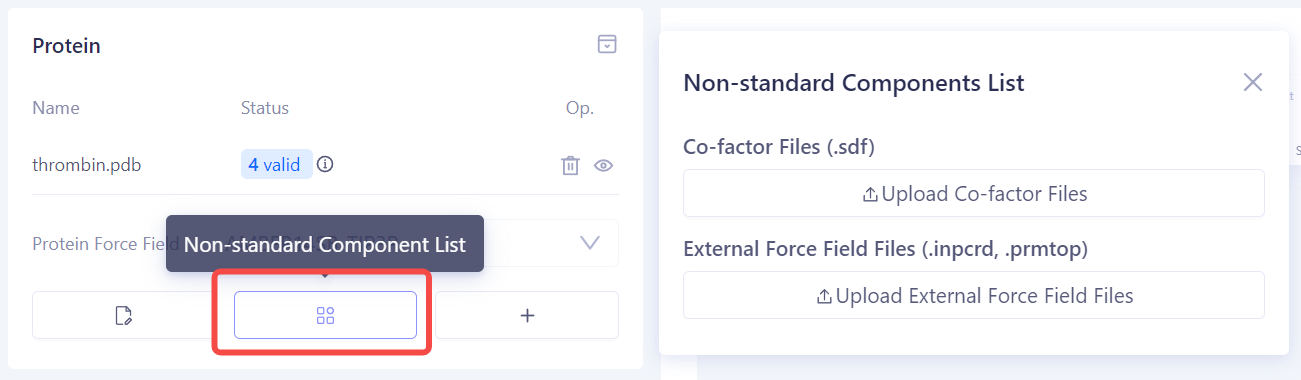
添加非标准组分:如果蛋白质结合位点附近存在非标准组分(例如辅助因子或金属离子),请在 Non-standard Components List 中上传相关的配体结构或构建好的力场参数文件,以确保模拟的准确性。 |
|
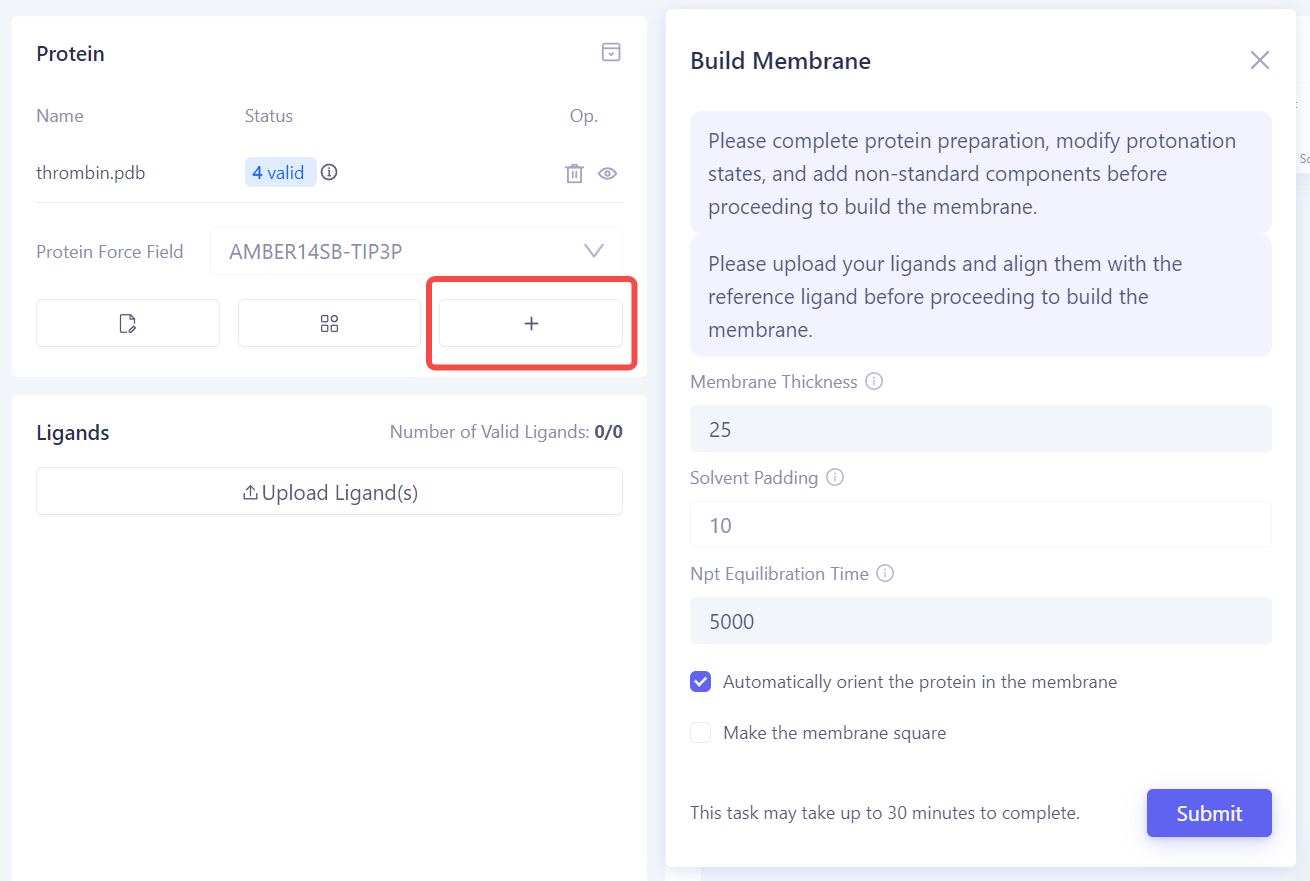
构建膜环境:如果目标蛋白是膜蛋白,请点击 Build Membrane 进入膜构建界面。在该界面中,您可以调整膜的厚度、溶剂填充层以及平衡时间等参数,确保蛋白正确嵌入膜中。 |
|
| 折叠蛋白窗格:完成上述步骤后,可以点击折叠按钮来隐藏蛋白设置的相关组件以节约屏幕空间。 |
|


| 小贴士:蛋白合法性检查的项目及可能的状态
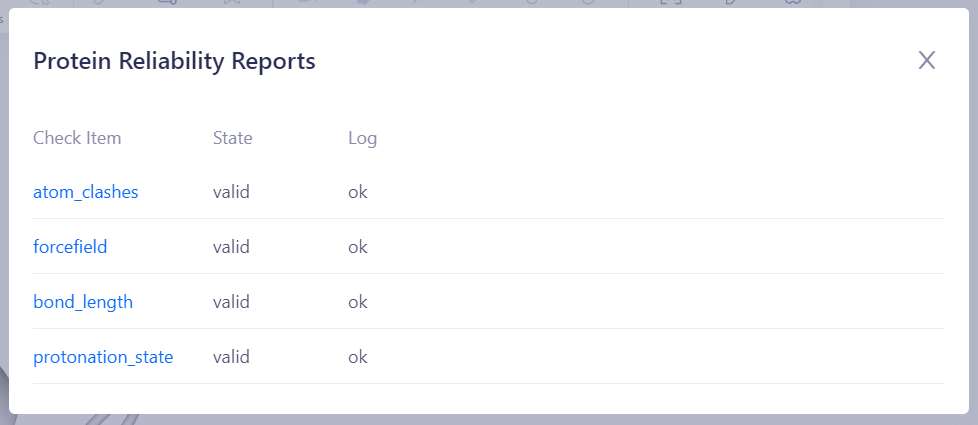
碰撞检查(Valid/Error):检测蛋白结构中是否存在原子间的不合理碰撞,常见问题包括原子重叠、非物理性接近、可替换残基异常、金属离子未配位等,这些问题可能导致模拟不稳定或能量极小化失败。 键长检查(Valid/Error):验证蛋白质中所有化学键的长度是否在合理范围内,避免异常的拉伸或压缩,从而确保分子动力学模拟的稳定性。 主链二面角检查(Valid/Warning):检查蛋白质主链的二面角是否位于合理的构象空间(如拉马钱德兰图的允许区域),避免出现不自然的主链构象,影响结构稳定性。 共平面检查(Valid/Warning):验证特定原子平面上的四面体中心原子是否存在不合理的扭曲或偏离共面现象,尤其在芳香环、共轭平面或特定氨基酸残基(如脯氨酸)中,这些异常可能影响能量计算的准确性。 力场参数检查(Valid/Error):确保蛋白质结构中的所有组分均具备完整且合适的力场参数,以便在后续分子动力学模拟中准确计算相互作用能量。 质子化状态检查(Valid/Warning):确保蛋白质中的氨基酸残基和配体的质子化状态在指定 pH 条件下是合理的,尤其是活性位点残基(如组氨酸、谷氨酸、天冬氨酸等),避免电荷分布错误影响结合自由能计算。 |
3.2.3 上传配体分子并进行分子对齐
配体准备是进行 FEP 计算的重要前置步骤,确保配体分子结构的合法性、准确性和与参考配体的空间叠合良好。本小节将分步骤指导您如何进行配体的上传、检查、叠合和实验数据的导入。
| 操作描述 | 界面截图 |
上传配体分子结构文件:在页面左上角的 配体窗格 中,点击 Upload Ligand(s) 按钮,选择要上传的配体分子结构文件(例如 1a.sdf)。系统会自动进行配体的合法性检查。如果上传的 .sdf 文件中包含多个配体,系统将自动将其拆分为多个独立的配体条目。 |
|
| 检查配体合法性:配体检查完成后,系统将显示检查结果状态,包括 "Valid"(有效)、"Warning"(警告)或 "Error"(错误)。您可以根据提示进行修正或进一步检查。 |
|
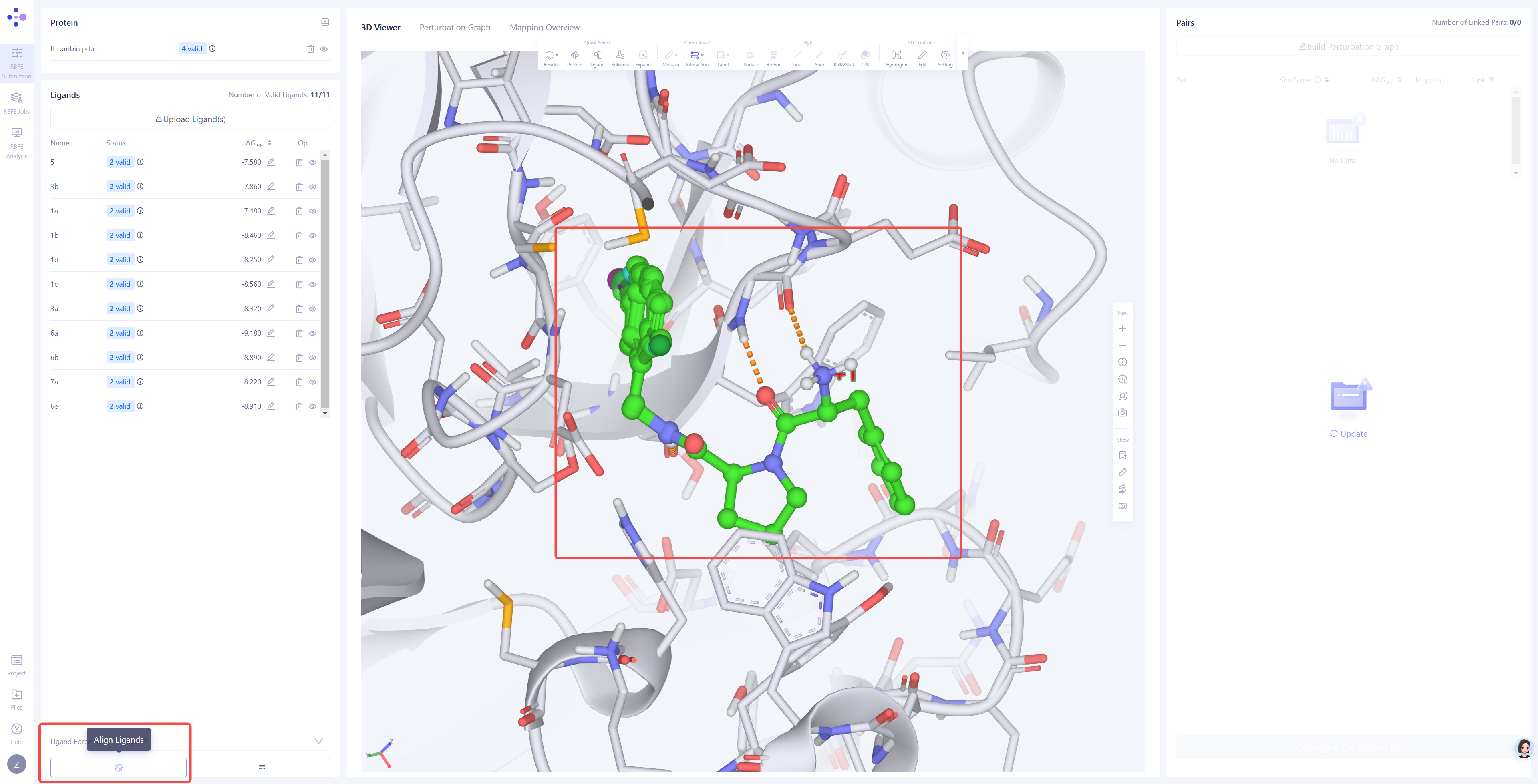
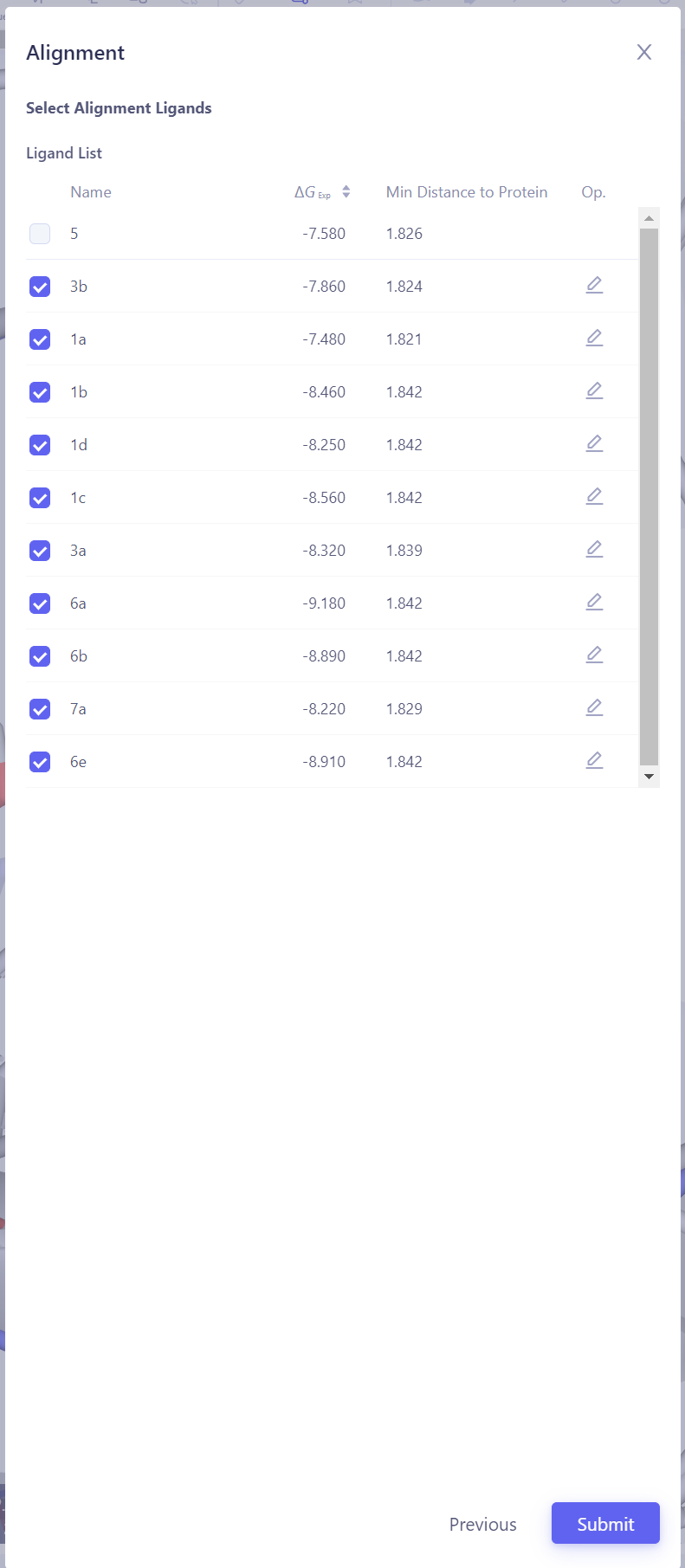
| 配体分子叠合:在 3D 窗格 中,检查上传的配体是否已经与参考配体分子良好叠合。如果未能正确叠合,请点击右下角的 "Align Ligands" 进行分子叠合操作。
选择参考分子:选择一个已与蛋白形成可靠相互作用的参考分子结构,确保其能够作为叠合的基础。然后点击 Next。
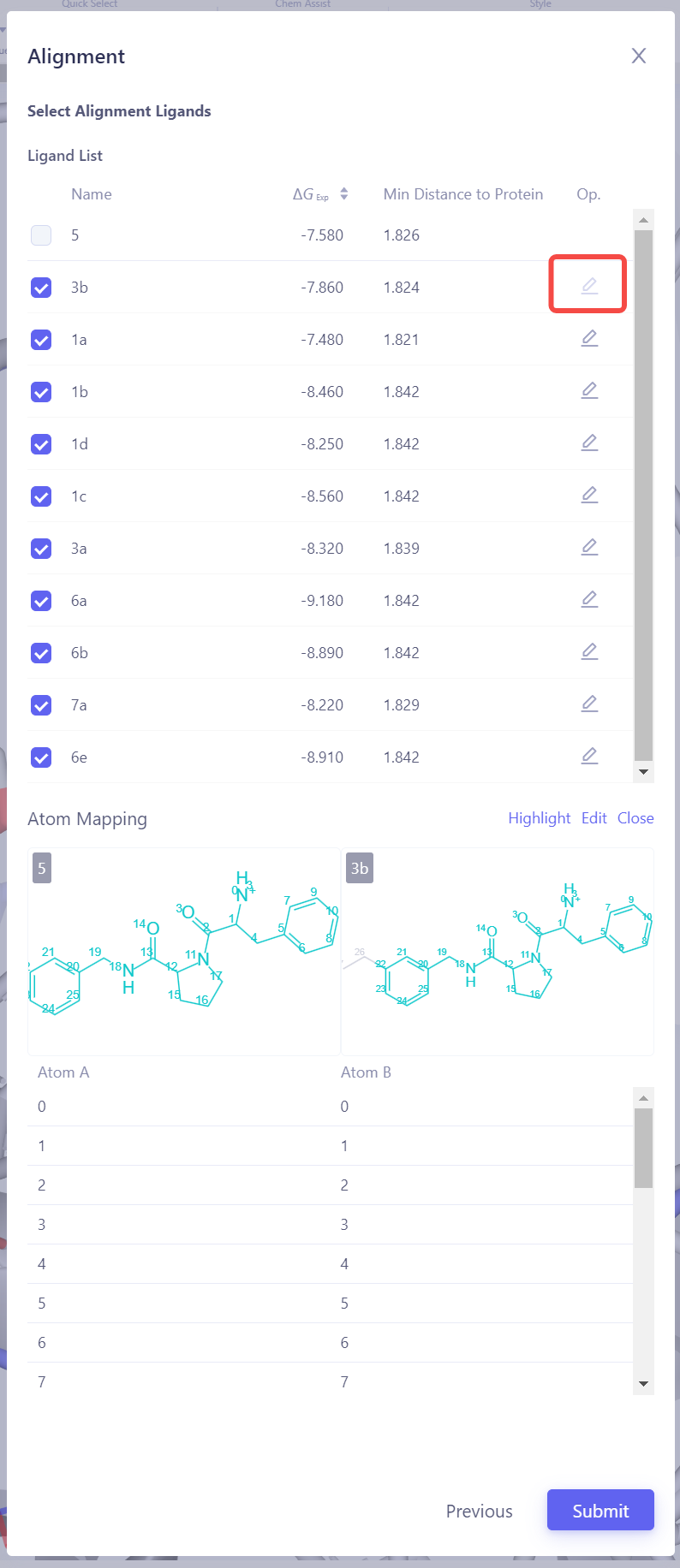
选择目标分子:选择需要与参考分子进行叠合的其他分子。点击 Atom Mapping 检查和确认原子映射的准确性,必要时进行手动修正。提交叠合任务:确认所有设置无误后,点击 Submit 开始分子叠合任务。验证叠合结果:等待叠合任务完成后,在 3D 窗格中检查所有配体的叠合效果。如果叠合效果不理想,可以尝试更换参考分子或调整原子映射设置,重新进行叠合,直至所有配体均达到良好的叠合效果。 |
|
上传实验活性数据:如果配体分子具有已测定的实验活性数据(例如 IC₅₀ 或 Ki 值),可以直接在系统中编辑相应分子的实验值,或点击右下角的 Upload Affinity Data,通过模板文件一键批量上传实验数据,确保实验值与配体分子正确关联。 |
|

| 小贴士:配体分子合法性检查的项目及可能的状态
键级检查(Valid/Error):检查配体分子中所有化学键的键级是否合理,确保化学结构的正确性,避免出现键序错误或非物理性的化学键。 碰撞检查(Valid/Error):检测配体分子内部是否存在原子间的不合理碰撞,或配体与蛋白质之间存在未解决的碰撞问题,这可能导致模拟不稳定或能量计算异常。 力场参数检查(Valid/Error):确保配体的每个原子类型都被正确识别,并分配了合适的力场参数,包括键长、键角、二面角以及电荷分布,以保证能量计算的准确性。 质子化状态检查(Valid/Warning):验证配体在指定 pH 条件下的质子化状态是否合理,特别是酸性和碱性基团,确保电荷分布准确无误,避免在模拟中引入非物理性电荷。 配体-蛋白距离检查(Valid/Warning):检查配体与蛋白质之间的空间距离,确保配体处于合理的结合位点,避免非物理性接近或过远距离,这可能导致结合自由能预测偏差。 |
3.2.4 构建微扰计算图并检查待计算分子对的原子映射
微扰计算图(Perturbation Graph)的构建和原子映射检查是 FEP 计算前的重要步骤,确保分子对之间的连接合理,原子映射准确无误。本小节将分步骤指导您如何进行微扰图的构建和映射检查。
| 操作描述 | 界面截图 |
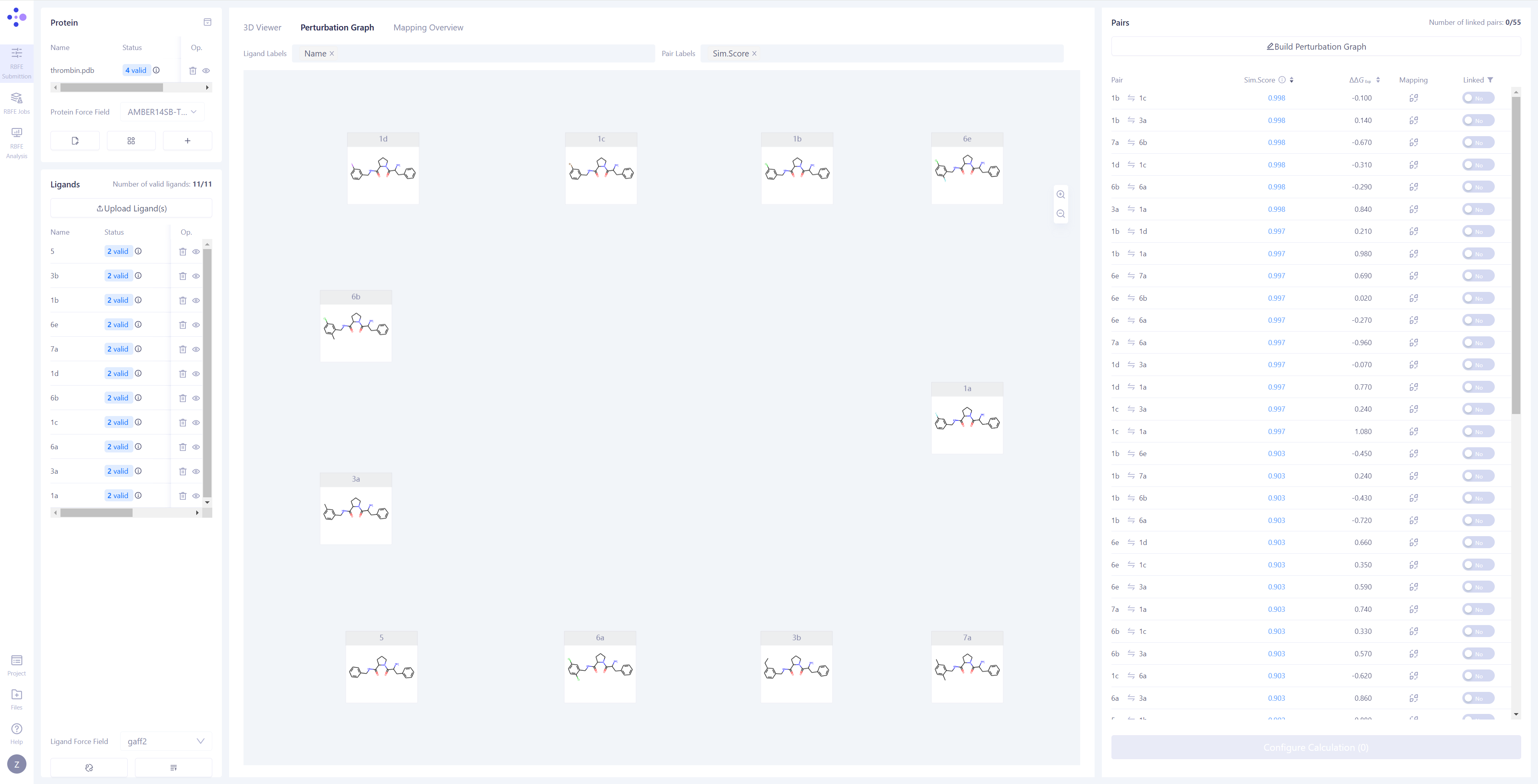
| 更新分子对信息:在右侧的 "分子对" 窗格中,点击 "Update" 按钮,系统将自动对所有被检测为 "Valid" 的分子进行两两 3D 原子映射,并将结果同步到表单中。该过程大约需要 1 分钟。 |
|
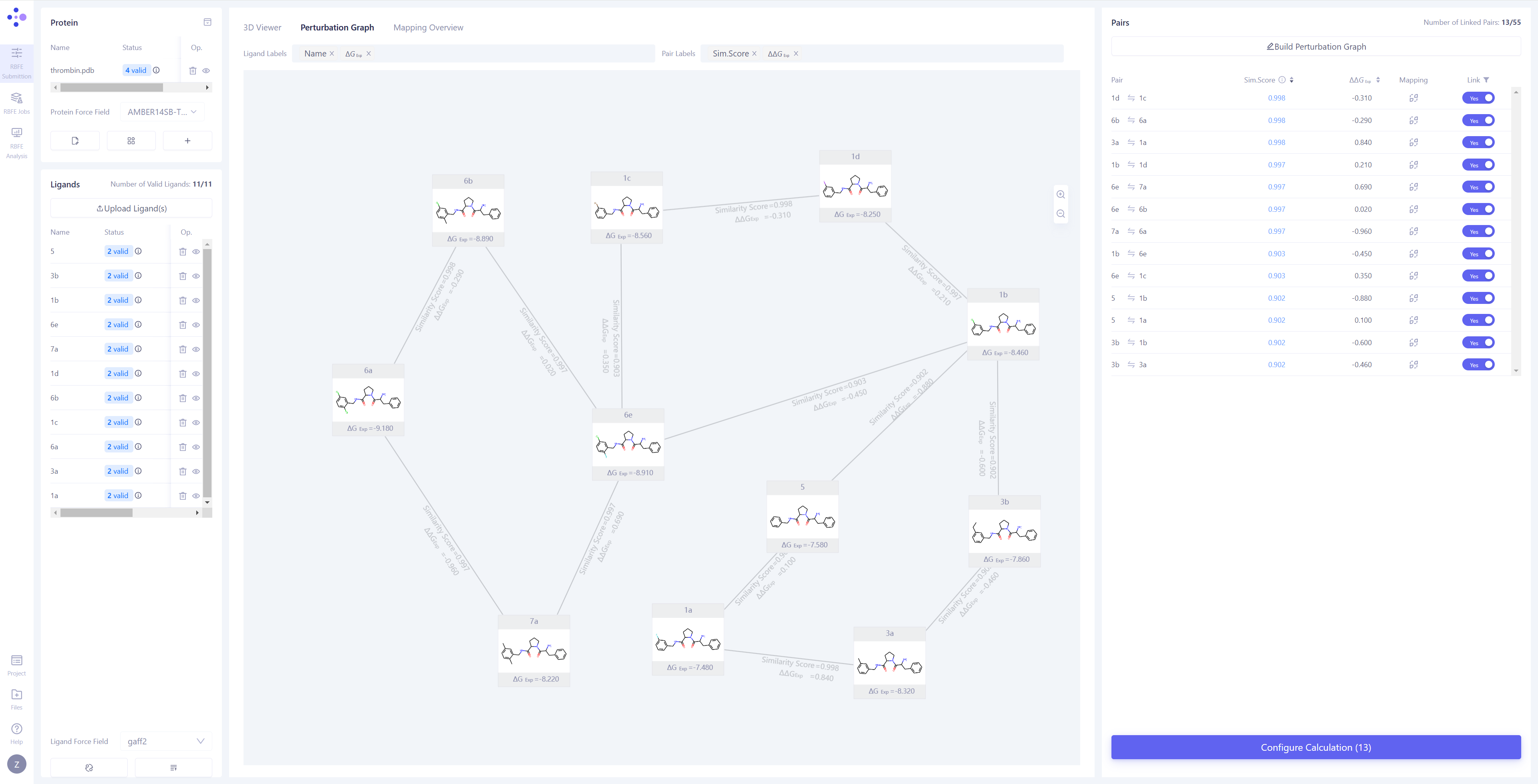
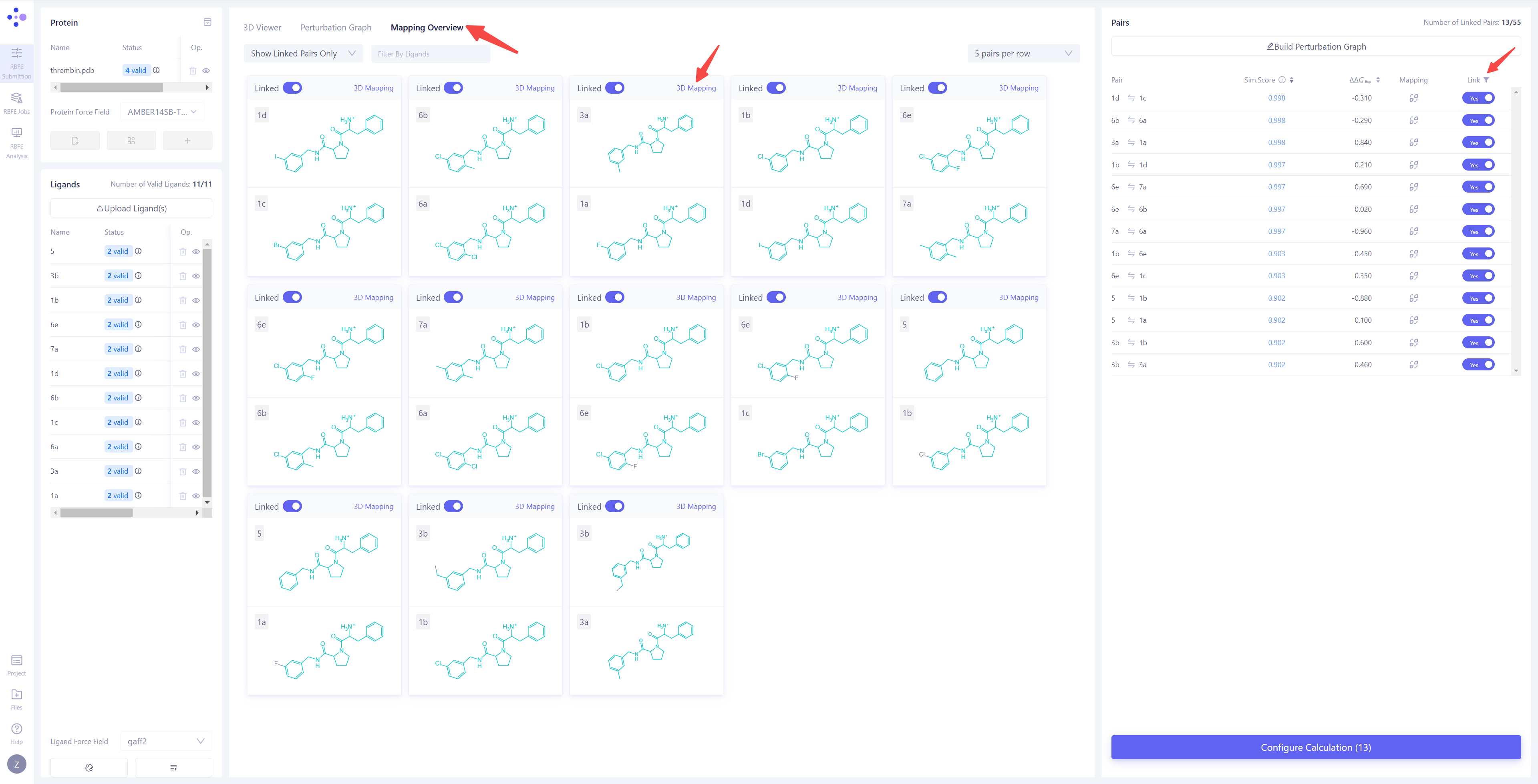
| 构建微扰计算图:同步完成后,中间视图将自动切换到 "微扰计算图 (Perturbation Graph)" 页面。您可以选择手动连接想要计算的微扰对,或通过 "Build Perturbation Graph" 自动构建微扰图。 自动构建微扰计算图:点击 "Build Perturbation Graph" 按钮,打开自动构建设置窗口,选择需要纳入计算的所有分子。如果希望构建星状微扰图(即将一个配体分子作为中心,与其他所有分子分别连接),请启用 "Build star-shaped perturbation graph" 选项,并选择中心配体。点击 "Submit" 按钮,系统将自动生成微扰计算图。 筛选已连接边:在右上角的 "Linked" 列表中,选择 "Yes" 选项,即可快速筛选并查看所有已连接的边及其相关信息。 |
|
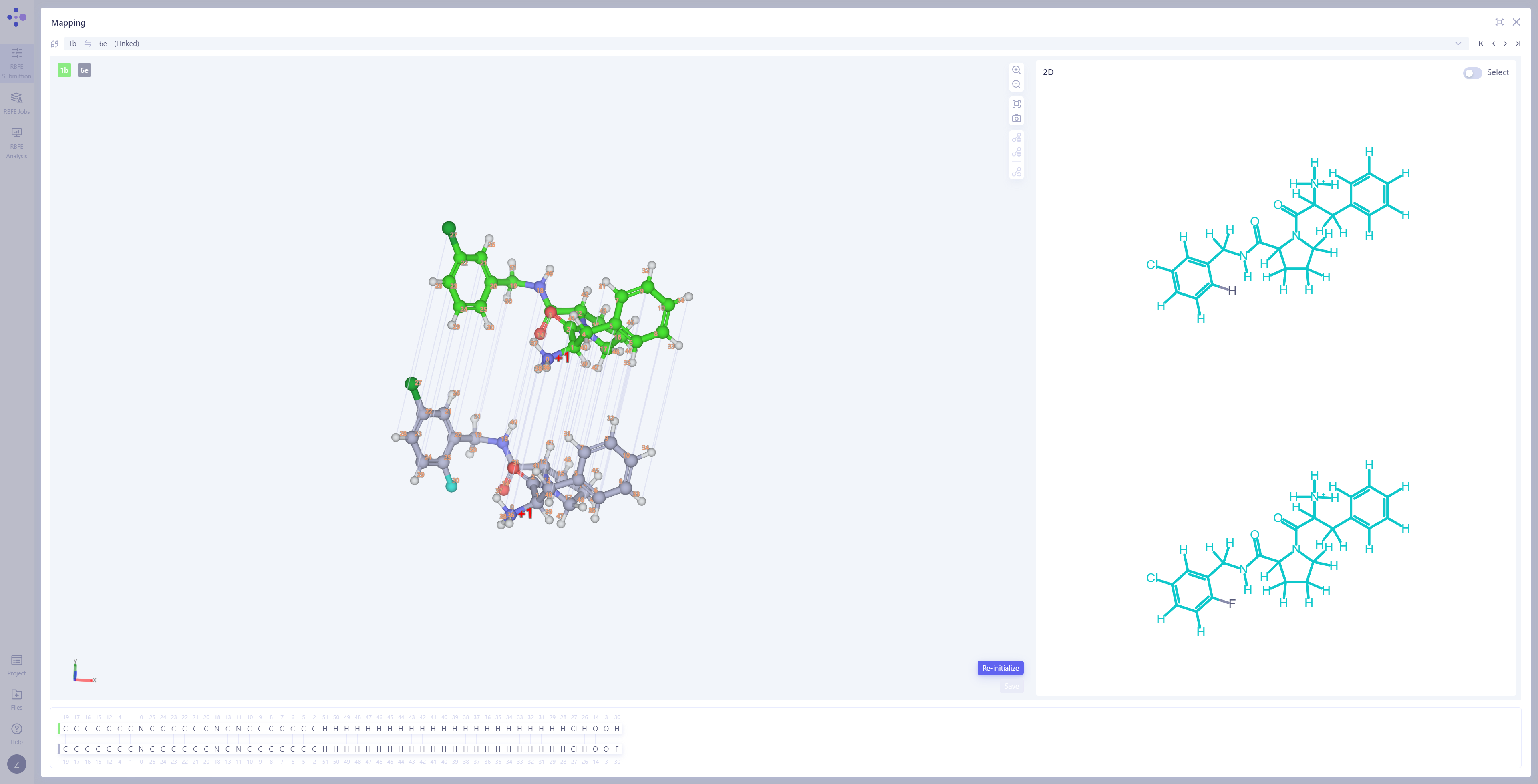
| 检查原子映射:将中间视图切换到 "Mapping Overview" 模式,检查所有已连接边的原子映射,确保映射的准确性和完整性。 打开原子映射详情:如果发现存在错误映射,或需要对特定原子对进行进一步检查,请点击 "3D Mapping" 打开原子映射详情界面。 修改原子映射:在 3D Mapping 窗格中,您可以:直接在 3D 视图 中选中原子对,进行原子映射的 添加 或 删除;在右侧的 2D 映射视图 中,通过框选进行批量映射操作;在下方 原子编号映射 区域,通过编号划选进行大规模调整。完成所有操作后,点击"Save"来保存修改后的原子映射。 |
|
3.2.5 设定计算参数并提交计算任务
| 操作描述 | 界面截图 |
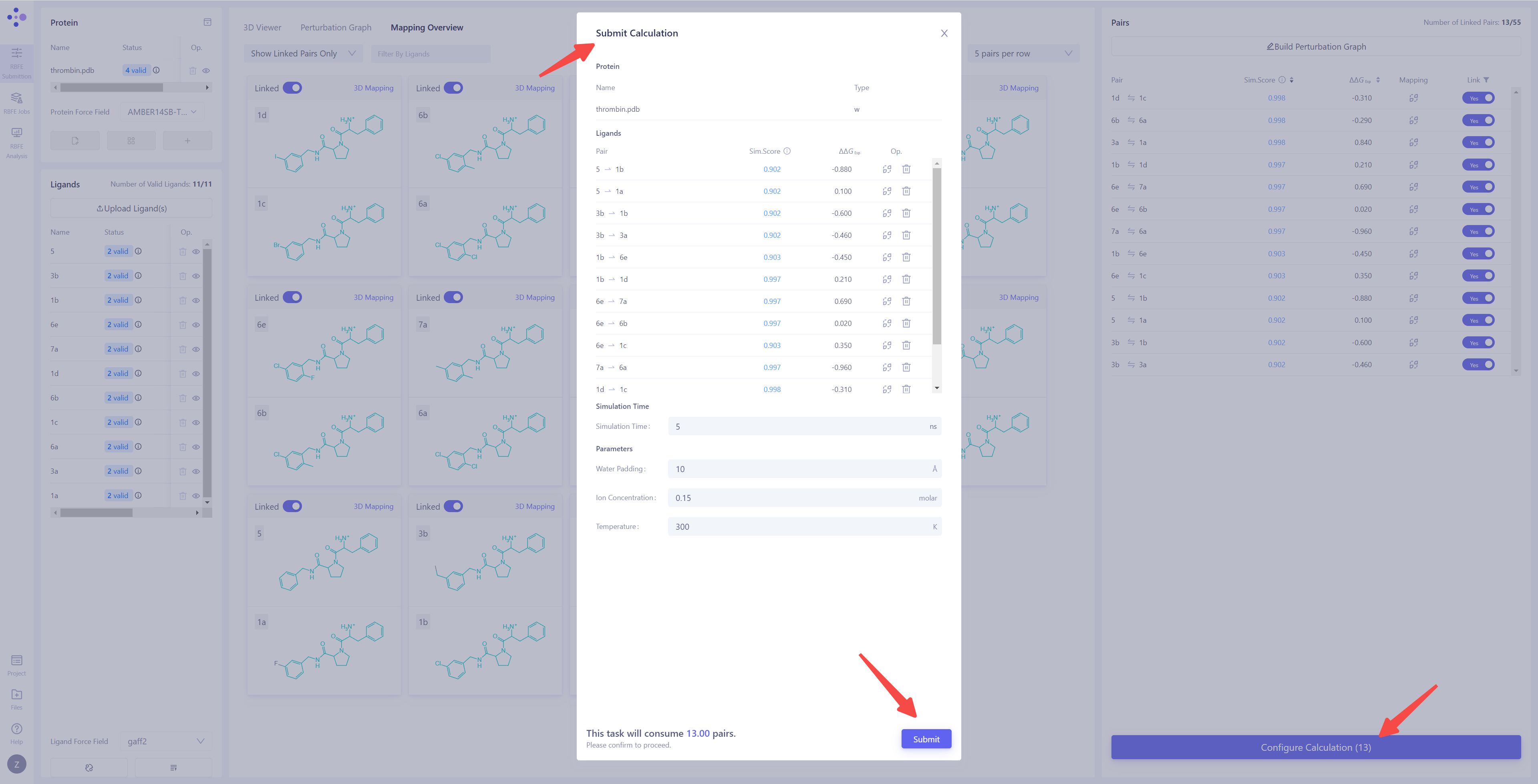
| 进入计算参数配置界面:当所有前置任务完成后,右下角的 "Configure Calculation" 按钮将变为可点击状态。点击该按钮进入 FEP 计算参数配置 界面,开始进行任务的详细设置。 确认膜蛋白体系标识:如果目标体系是 膜蛋白,请确保 "Type" 列中已标记为 "with Membrane",以保证模拟条件正确匹配膜环境。 检查微扰对:确保所有微扰对符合预期。可以点击 "Mapping" 进行二次原子映射检查,确保所有映射正确无误。如果存在不需要计算的微扰对,可以选择对应条目,点击 "Delete" 将其移除。 设置模拟时长(Simulation Time):对于常见体系,建议将模拟时长设置为 5 ns,以确保计算结果的稳定性和准确性。 配置模拟参数:设置添加水分子的层厚(Water Padding),以确保体系边界不会影响模拟结果;设置离子浓度(Ion Concentration),离子为 Na⁺ 和 Cl⁻,用于中和体系电荷并模拟生理环境;设置模拟温度(Temperature),通常为 300 K,以匹配常规实验条件。 确认计算费用:完成所有参数设置后,系统将自动统计本批次 FEP 计算任务的预计费用。请仔细核对所有设置和计算资源信息,确保无误。 提交计算任务:点击 "Submit" 按钮,所有 FEP 计算任务将被提交到服务器,开始正式计算。 |
|
3.3 FEP 任务监控与结果分析
3.3.1 监测任务进程和状态
| 操作描述 | 界面截图 |
| 进入 FEP 任务管理界面:在左侧侧边栏中,点击 "RBFE Jobs",进入 FEP 任务管理 界面 页面左上角:展示了所有任务的 状态统计,包括已完成、进行中、失败等不同状态的任务数量。 页面右上角:选项按钮,允许用户切换分子对的 2D 结构图片展示,以及是否显示每个分子对的模拟信息,以便根据需求自定义界面视图。 |
|
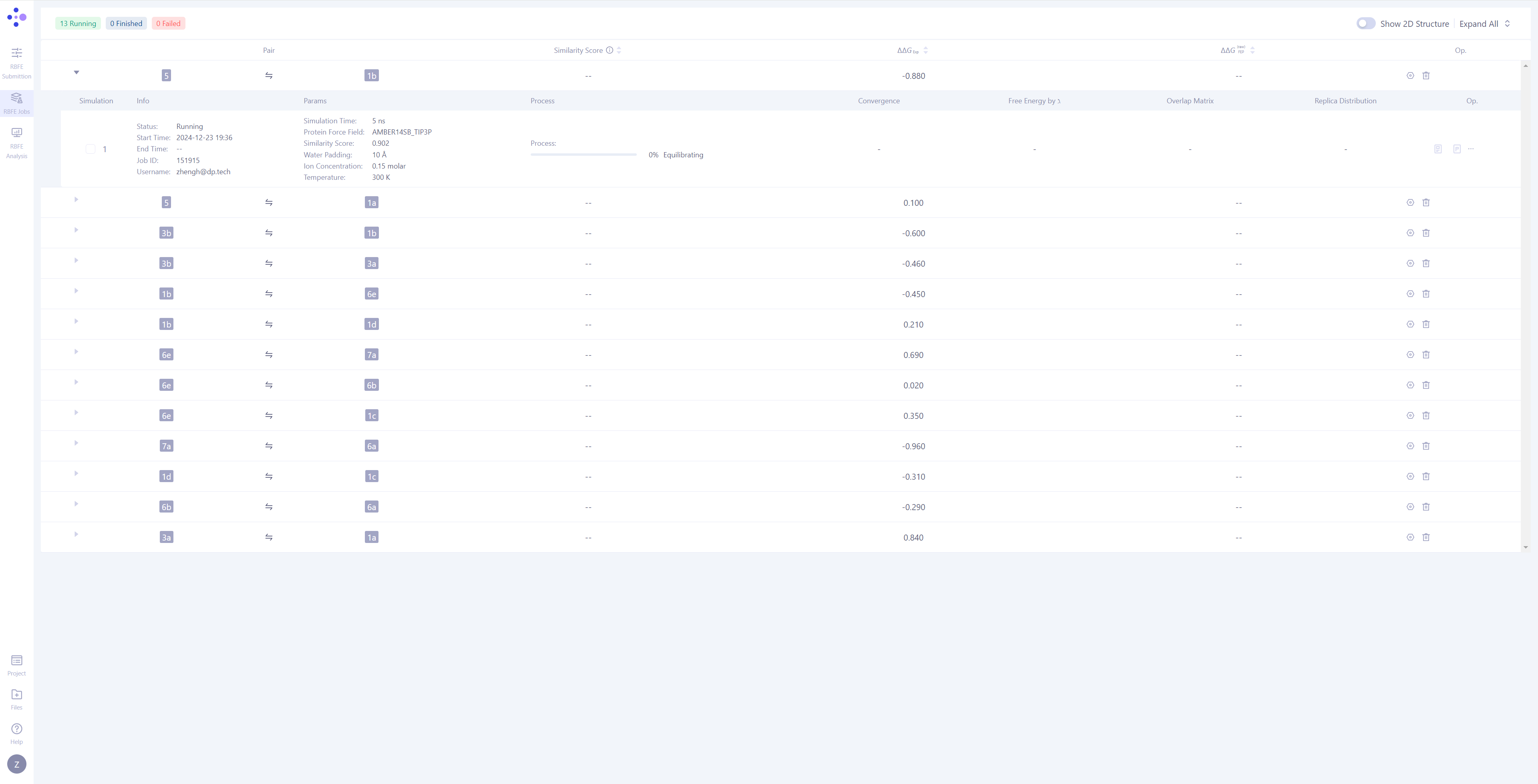
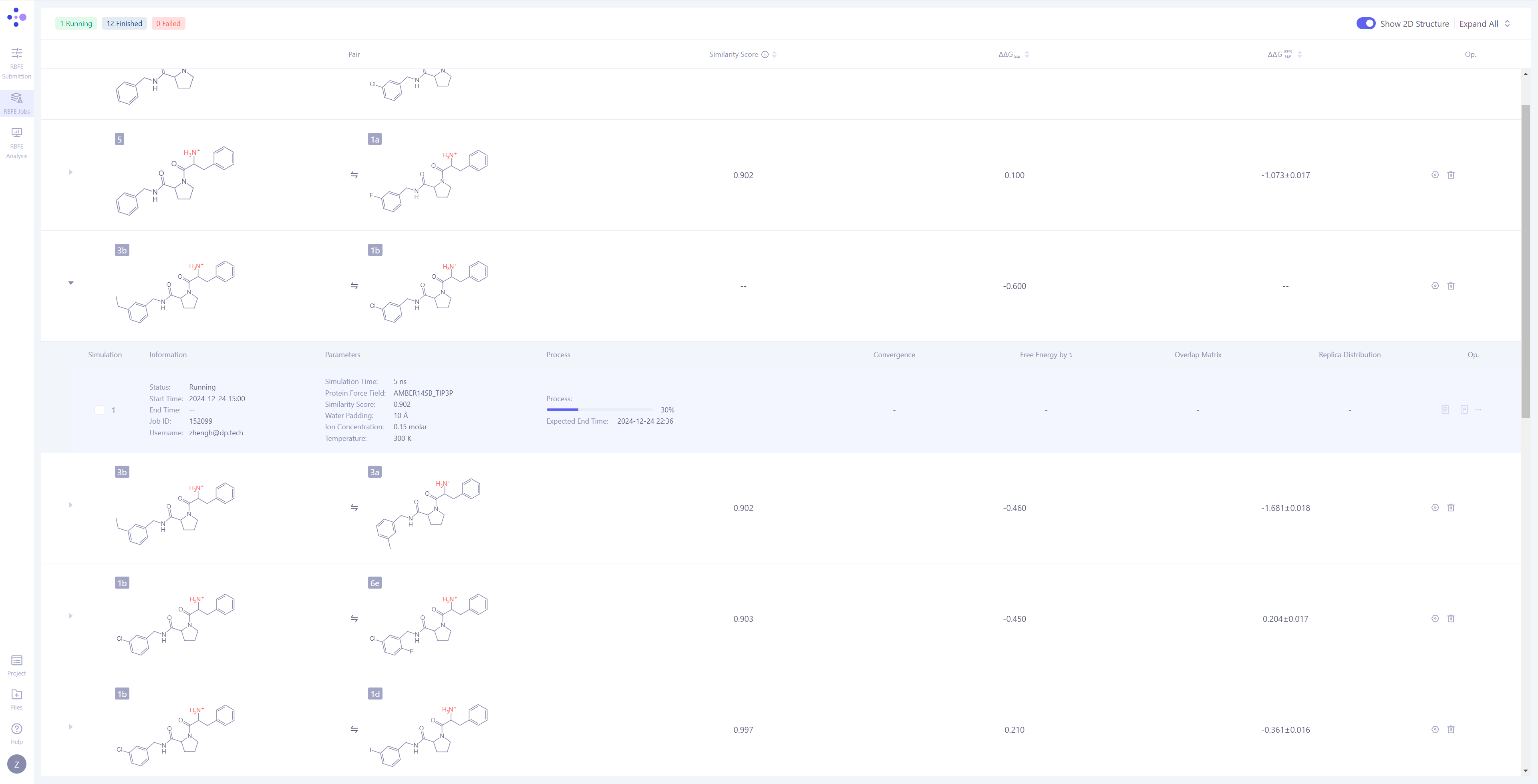
| 任务管理的分子对分组:在 Hermite 中,FEP 任务是以 分子对 作为分组进行管理的,每次独立的模拟任务是最小的管理单元。 展开任务详情:点击每个分子对左侧的 三角箭头,可以展开显示该分子对的详细模拟信息。展开分子对的模拟信息后,您可以查看包括基本信息、模拟参数、任务进度、结果图示,还有相对应的操作按钮。 监控任务进度:任务有等待(Waiting)、平衡(Equilibrating)、生产(Production)三种任务状态。对处于模拟阶段的 任务,可以观测其运行进度和预期结束时间。 |
|
3.3.2 分析单个分子对计算结果
单个分子对的 FEP 计算完��成后,可以通过一系列可视化分析工具对模拟结果进行详细解析,评估计算的准确性和可靠性。本小节将指导您如何解读和分析单个分子对的计算结果。
| 操作描述 | 界面截图 |
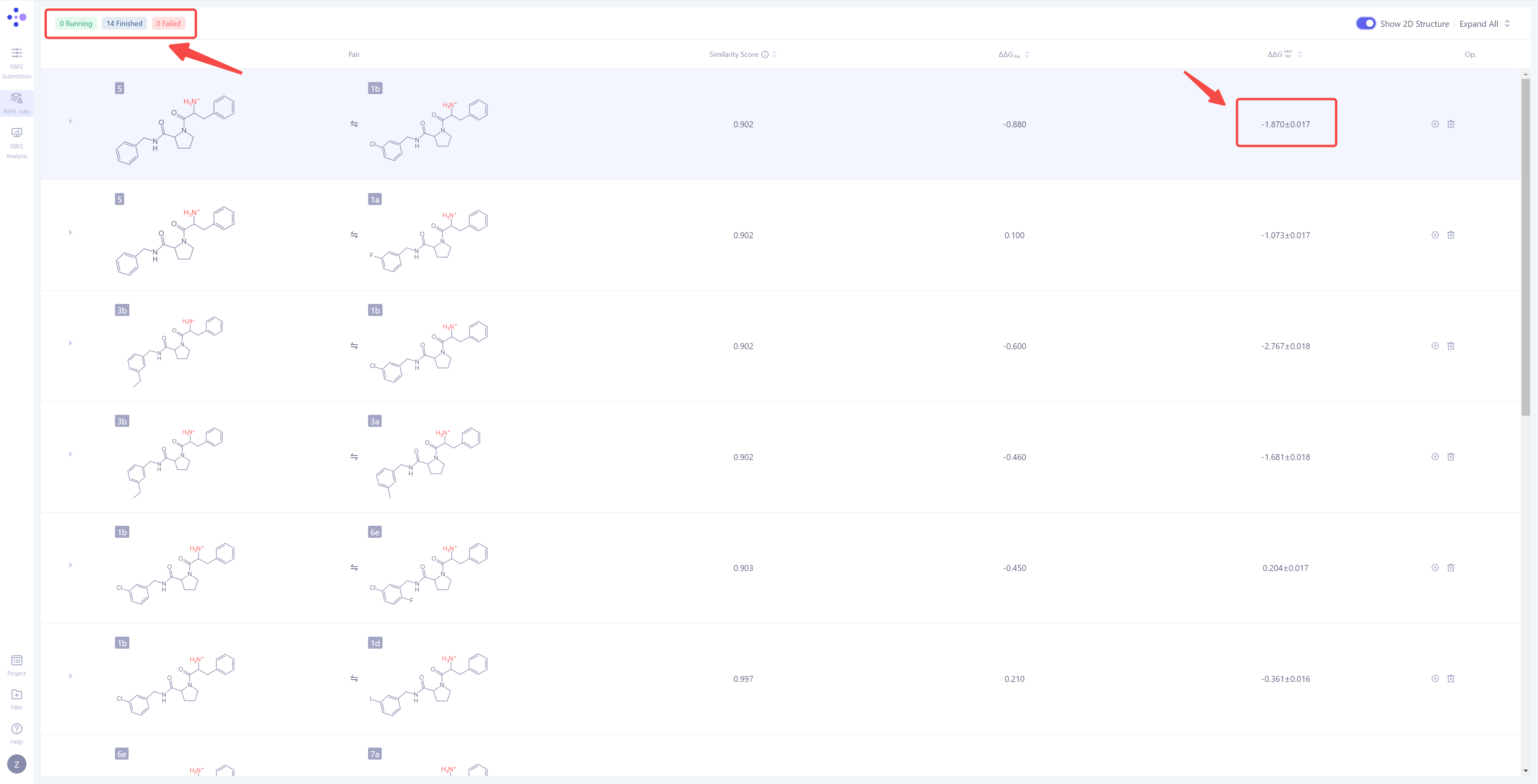
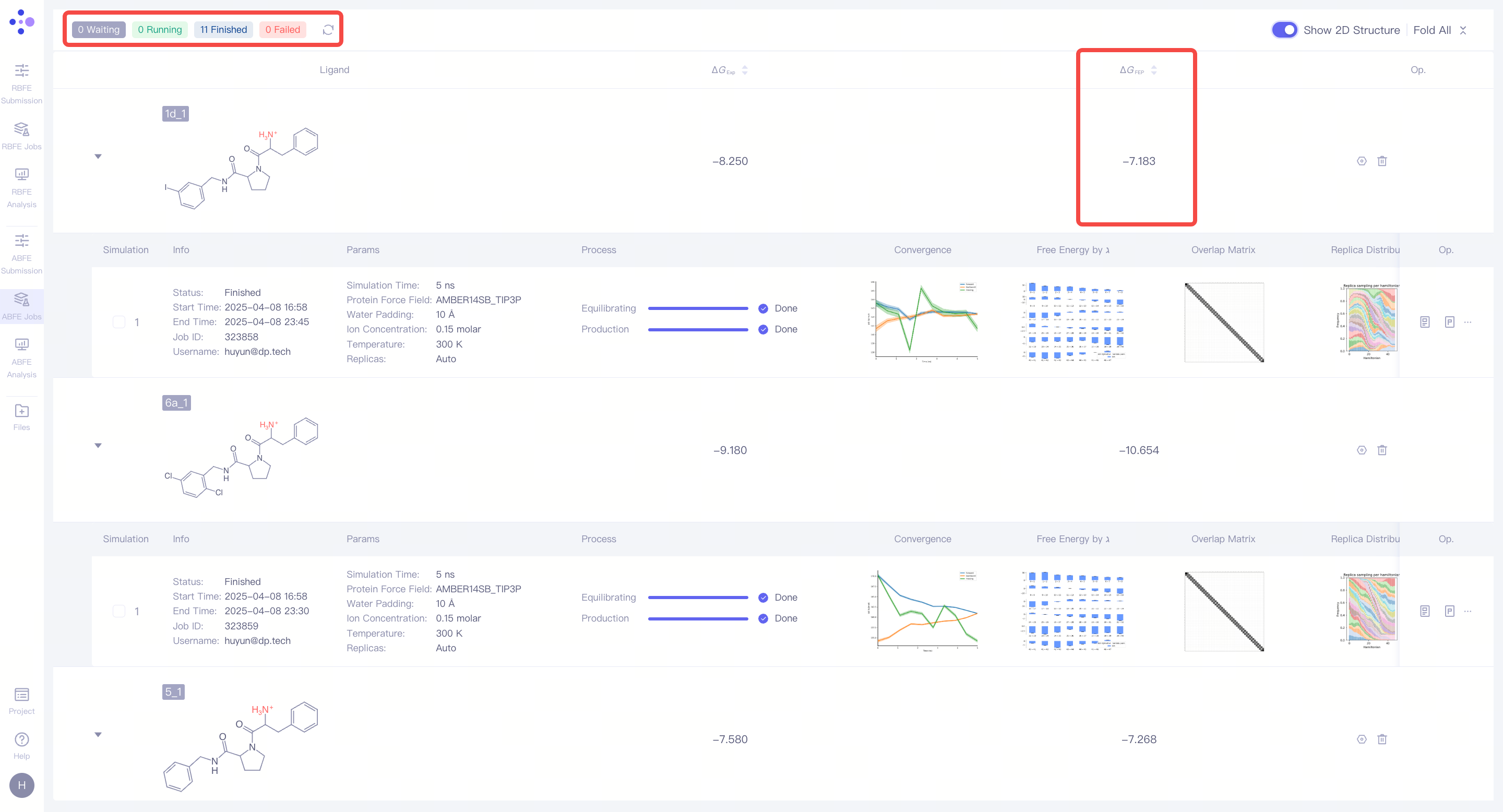
| 查看计算结果:当单个模拟任务成功完成后,左上角会显示 任务完成 状态,同时,该分子对的 ΔΔG_FEP 数值将自动计算并展示在界面上,作为分子对结合自由能差异的预测结果。 |
|
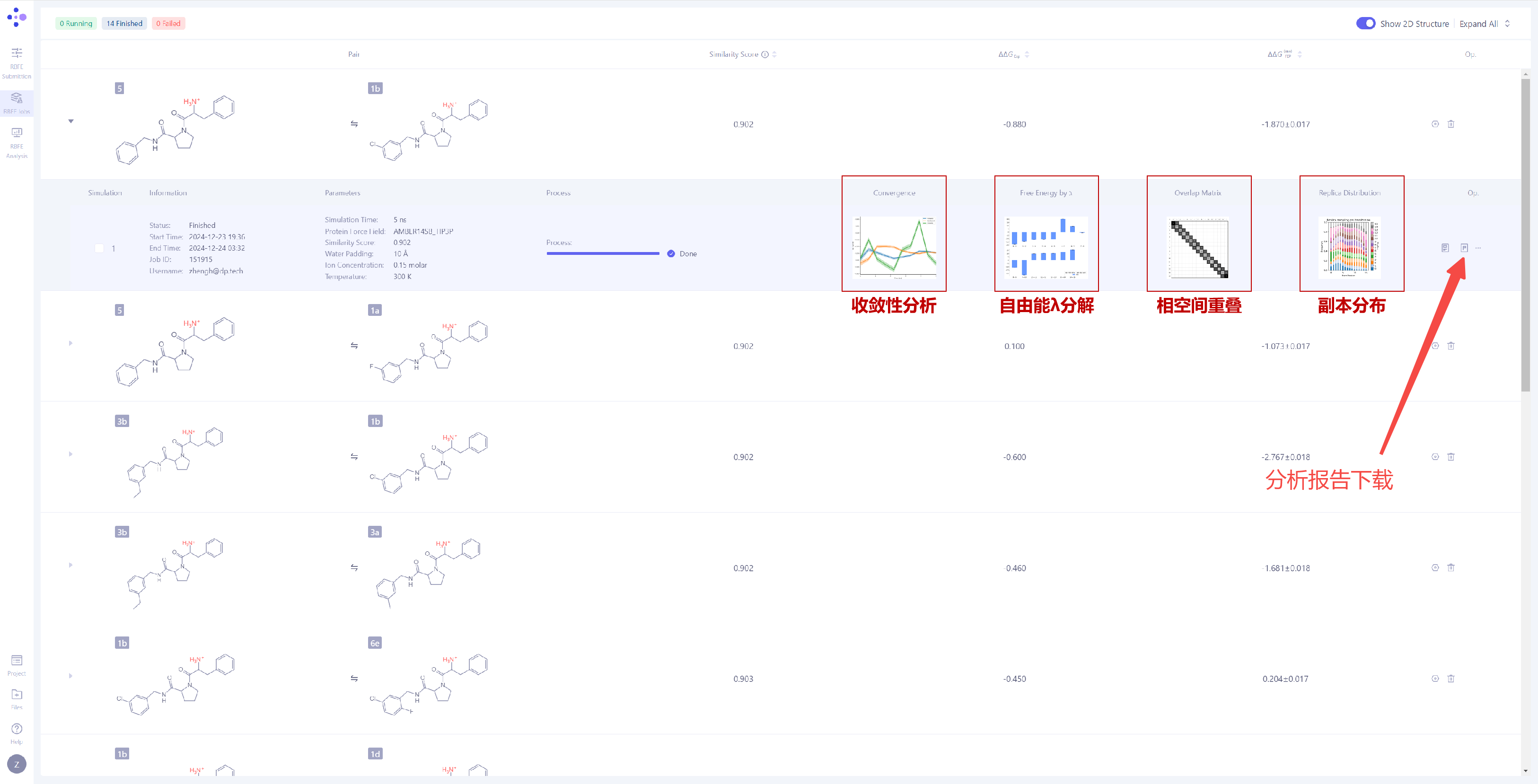
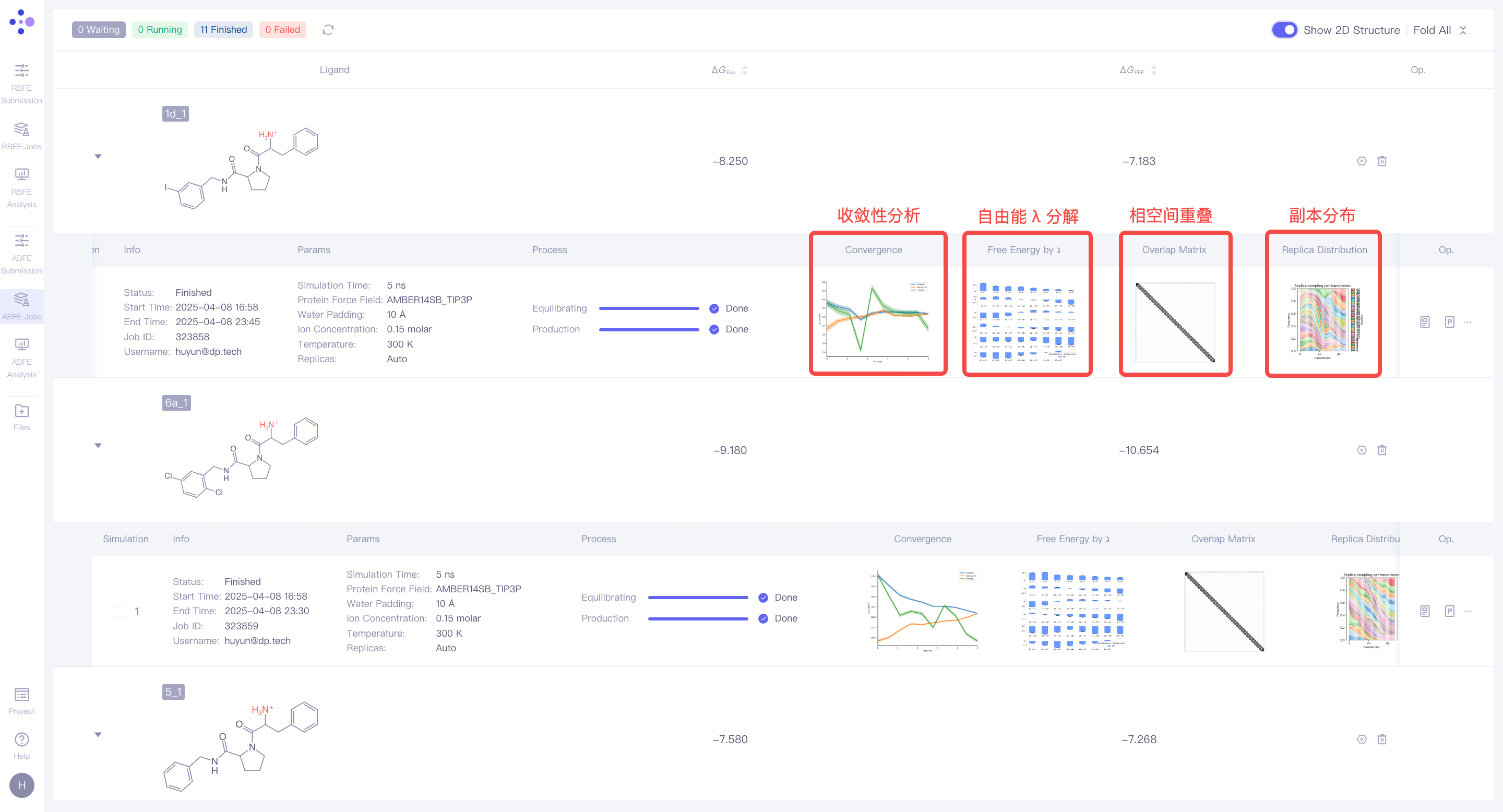
| 分析模拟收敛性:展开任务详情,可以观察本次 FEP 计算的 模拟收敛性,系统提供以下四种关键分析图表: 收敛性分析:收敛性分析展示了前向与后向自由能结果之间的差异,这种差异反映了滞后效应的程度。在理想情况下,前后向结果的差异应非常小,表明体系已经充分收敛,采样偏差可忽略不计。如果差异较大,通常意味着状态重叠不足或采样不充分。此外,平滑且稳定的移动平均值暗示自由能数值已趋于收敛,而持续变化的移动平均值则说明体系尚未完全收敛,仍在演化中。 自由能 λ 分解:自由能λ分解反映了体系在不同λ状态下的自由能变化情况。如果ΔΔG在λ区间内呈现平滑变化,说明体系在炼金转换过程中的过渡较为稳定,系统表现良好。若在某些λ值处出现突兀的跃变,可能暗示该阶段存在关键转换点,或采样不足,亦或是体系存在不稳定性。 相空间重叠:相空间重叠矩阵展示了不同λ状态之间的构象采样重叠度。每个单元格的数值代表两个相邻λ窗口之间的构象空间共享程度,数值越高(通常建议大于 0.15),表示相邻状态之间的过渡更平滑,计算结果也更可靠。如果某些区域的重叠度较低,可能表明状态之间存在显著的能垒或采样不足。 副本分布:副本分布展示了不同λ区间的采样覆盖情况。均匀分布意味着所有λ状态都得到了充分的采样,整个系统在所有窗口中的采样均匀且稳定。如果分布不均匀,可能暗示某些λ区间存在采样瓶颈或能量壁垒,影响最终自由能的准确性。 更详细的分析结果可以通过点击 "Report" 下载完整的报告文件进行深入解析。 |
|
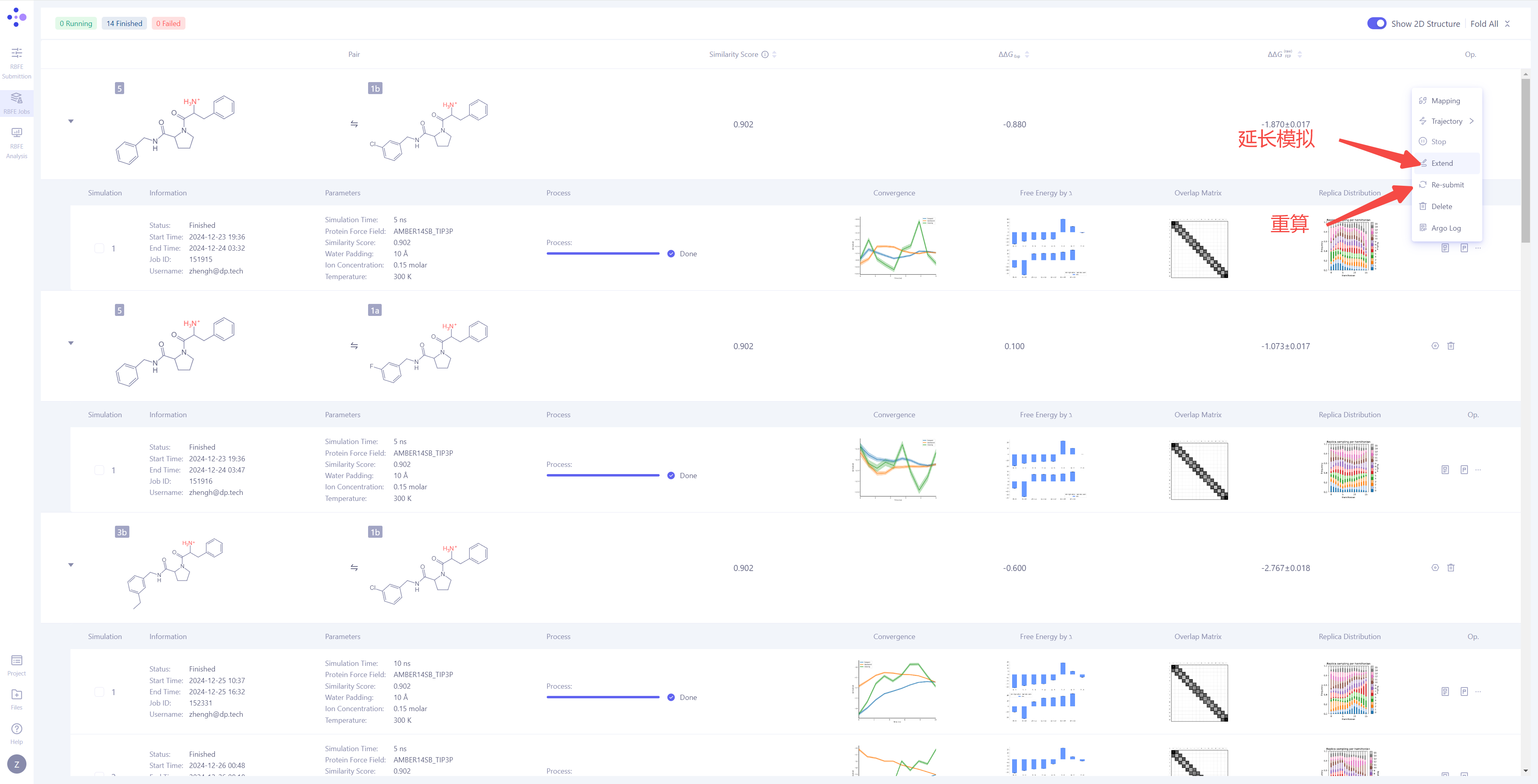
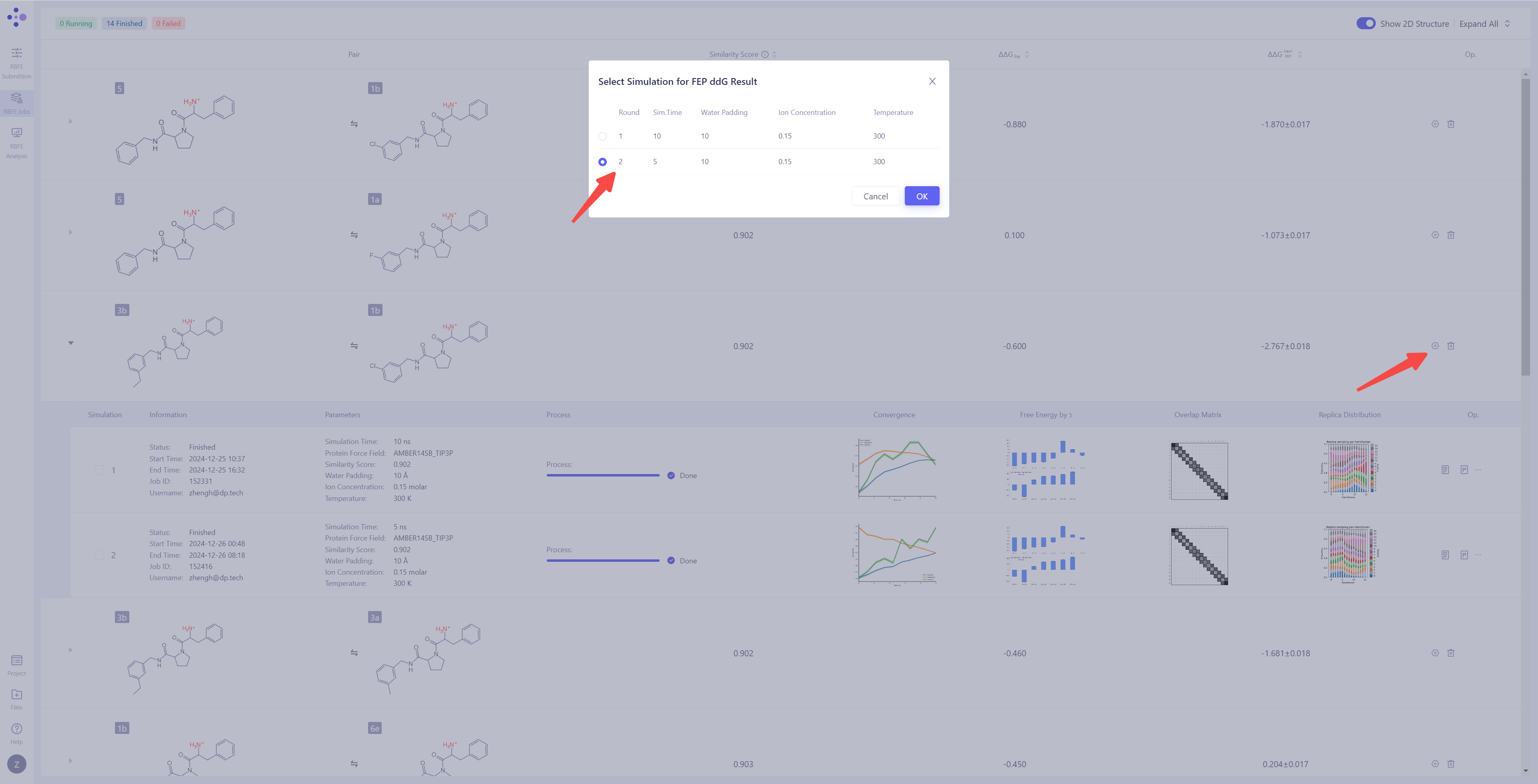
| 处理未收敛或不满意的结果:如果发现模拟结果未收敛或结果不理想,可以点击 "Extend" 按钮,延长模拟时间,增加采样量以提高结果的稳定性。如果需要重新进行计算,可以点击 "Re-submit" 按钮,重新启动该分子对的模拟任务。当一个分子对有多个模拟结果时,可以选择其中某一次特定的模拟作为最终的 ΔΔG_FEP 计算来源(系统默认选择最近一次的模拟结果)。 |
|
3.3.3 汇总所有分子对的计算结果
| 操作描述 | 界面截图 |
| 进入分析界面:当所有分子对的计算任务完成,并确认每对分子对的模拟结果已收敛后,点击左侧侧边栏的 "RBFE Analysis" 进入整体结果分析界面。在该界面中,用户可以基于热力学循环一致性、ΔΔG 相关性和ΔG 相关性进行系统分析。 | |
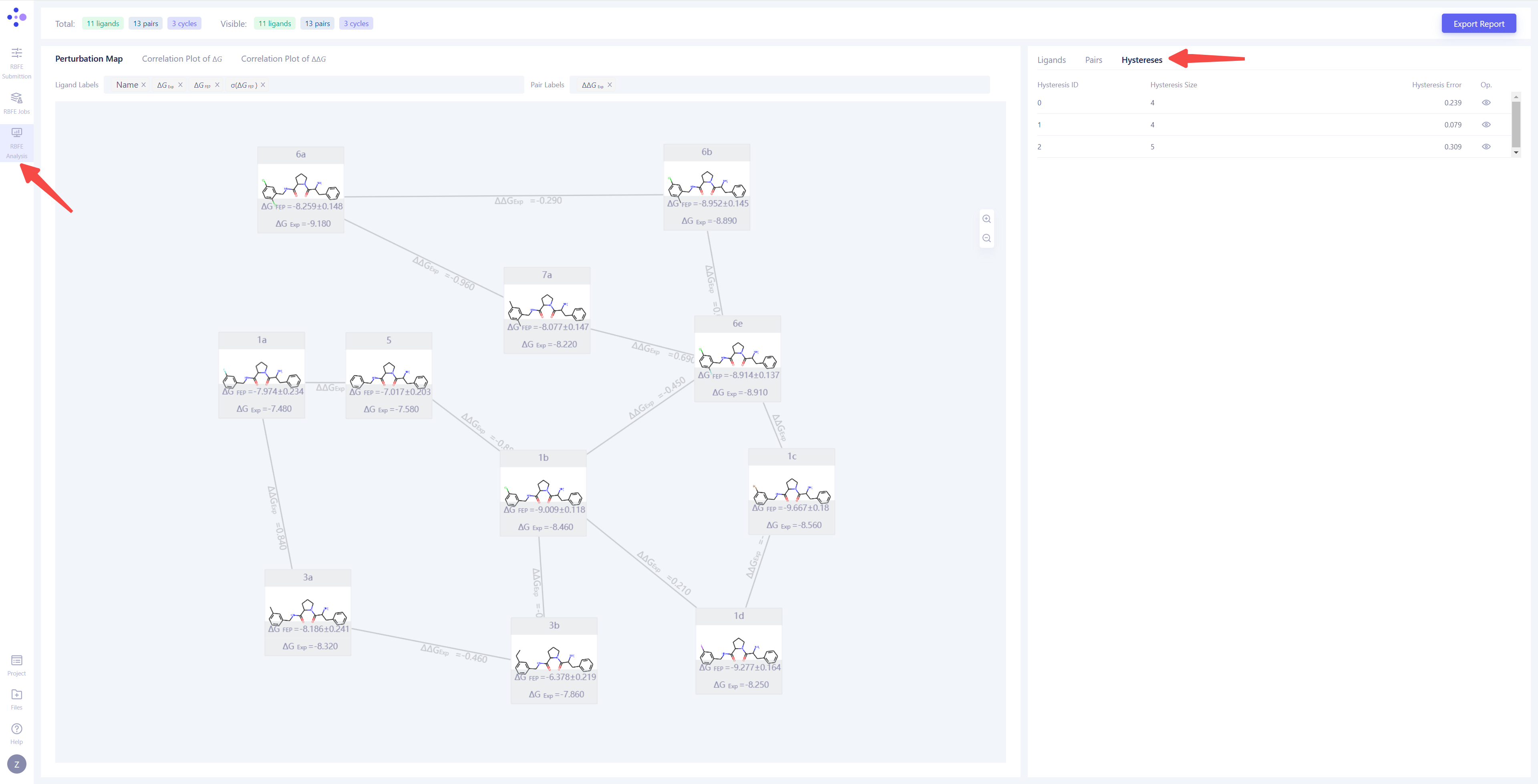
| 热力学循环一致性分析:首先,选择 "Hystereses" 视图,分析模拟结果的 热力学循环一致性。热力学循环误差(Hystereses Error)反映了热力学闭环的计算误差,理想情况下,该误差应接近 0。一般来说,当 Hystereses Error < 0.8 时,可以认为该热力学循环是可靠的。如果误差较大,建议检查热力学循环中涉及的分子对,延长不收敛分子对的模拟时长或重新进行计算。 |
|
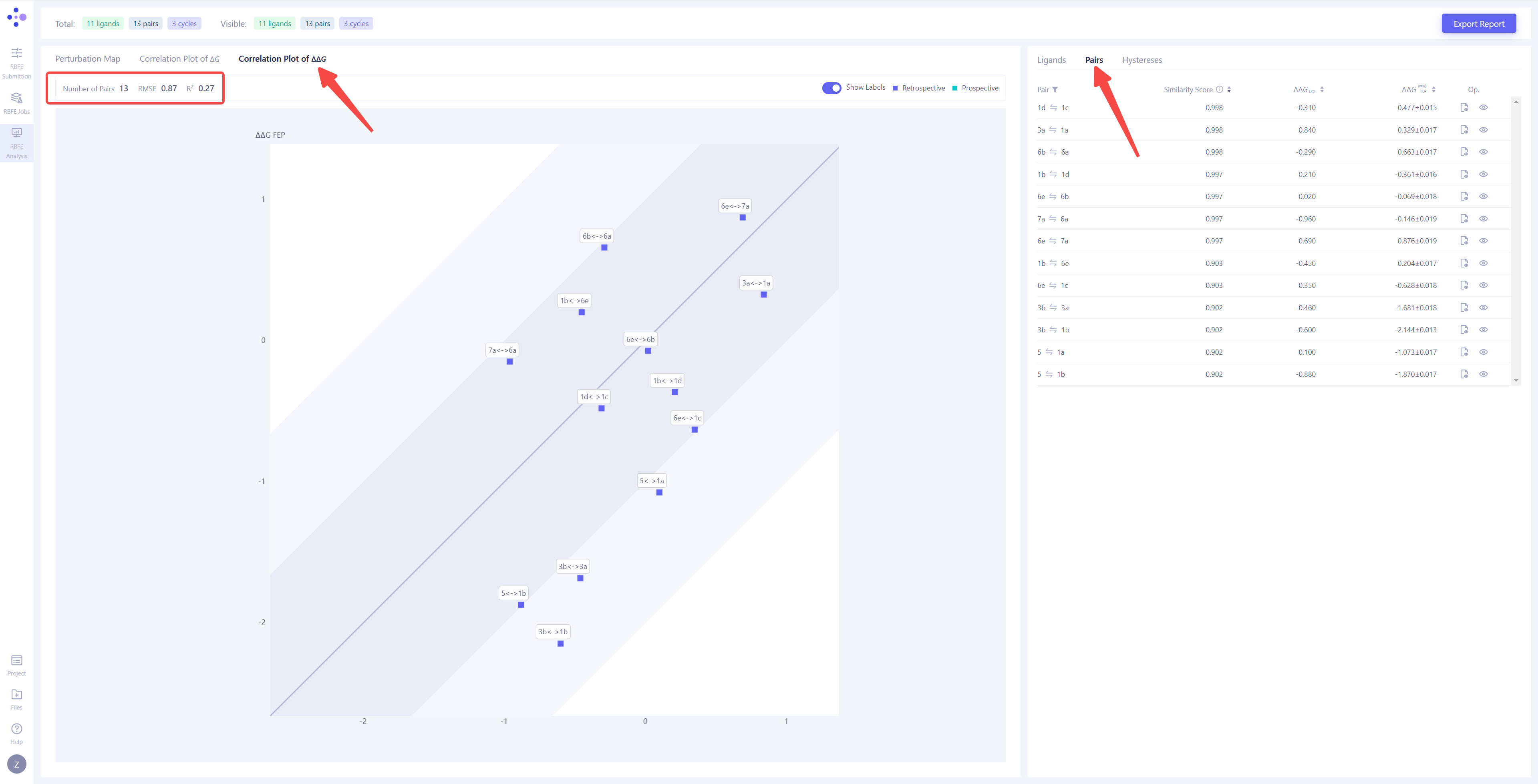
| 分子对计算结果相关性分析:将左视图切换到 "Correlation Plot of ΔΔG",右视图切换到 "Pairs",可以对每一对分子对的 FEP 计算结果进行分析。在相关性图中,横轴表示 ΔΔG 的实验值,纵轴表示 ΔΔG 的 FEP 计算值。深色区域表示 <1 kcal/mol 的误差范围;浅色区域表示 <2 kcal/mol 的误差范围。点越靠近对角线,说明计算结果越准确和可靠。如果有分子对落在 2 kcal/mol 的误差范围之外,建议重新进行 FEP 计算,以提高预测准确性。 |
|
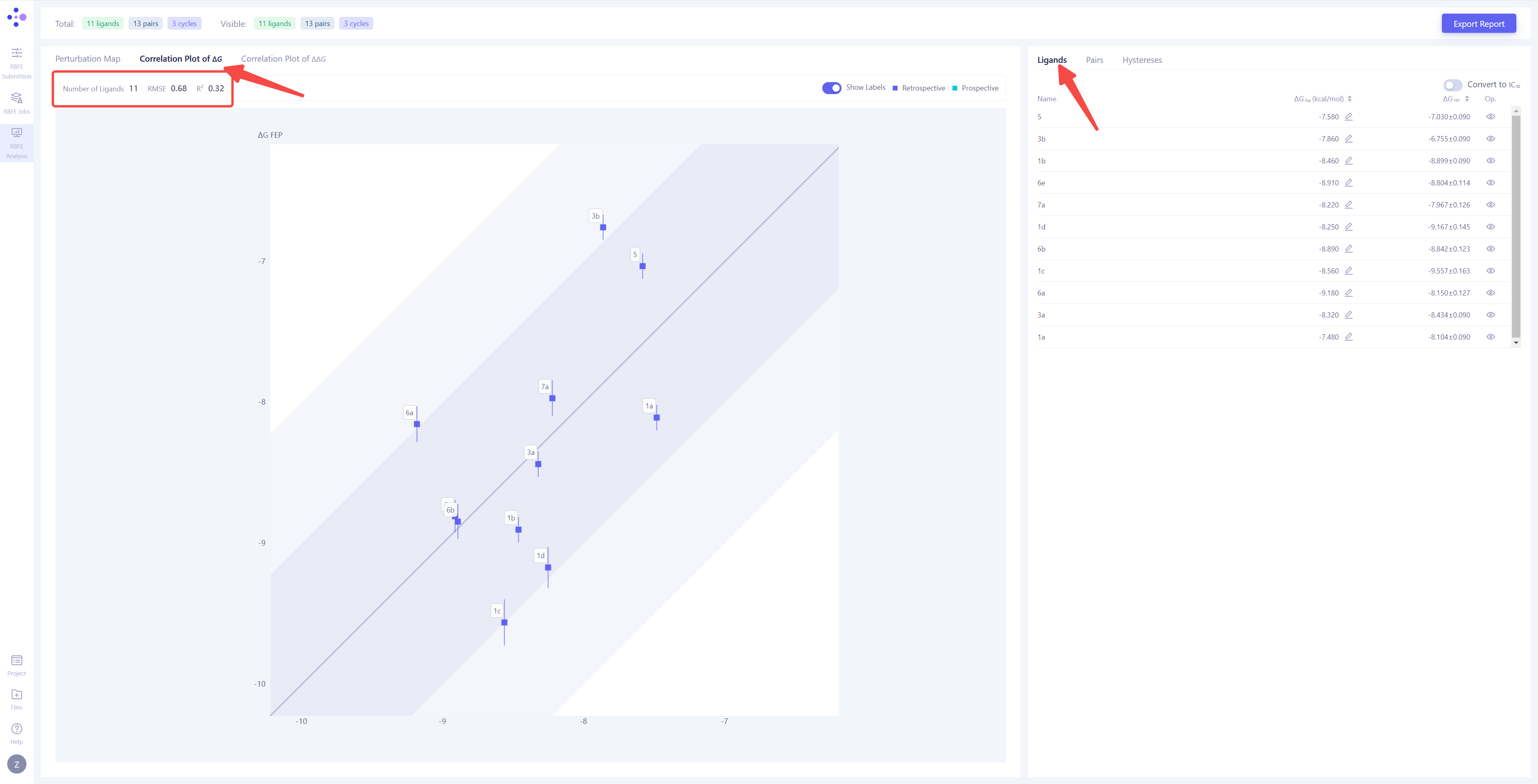
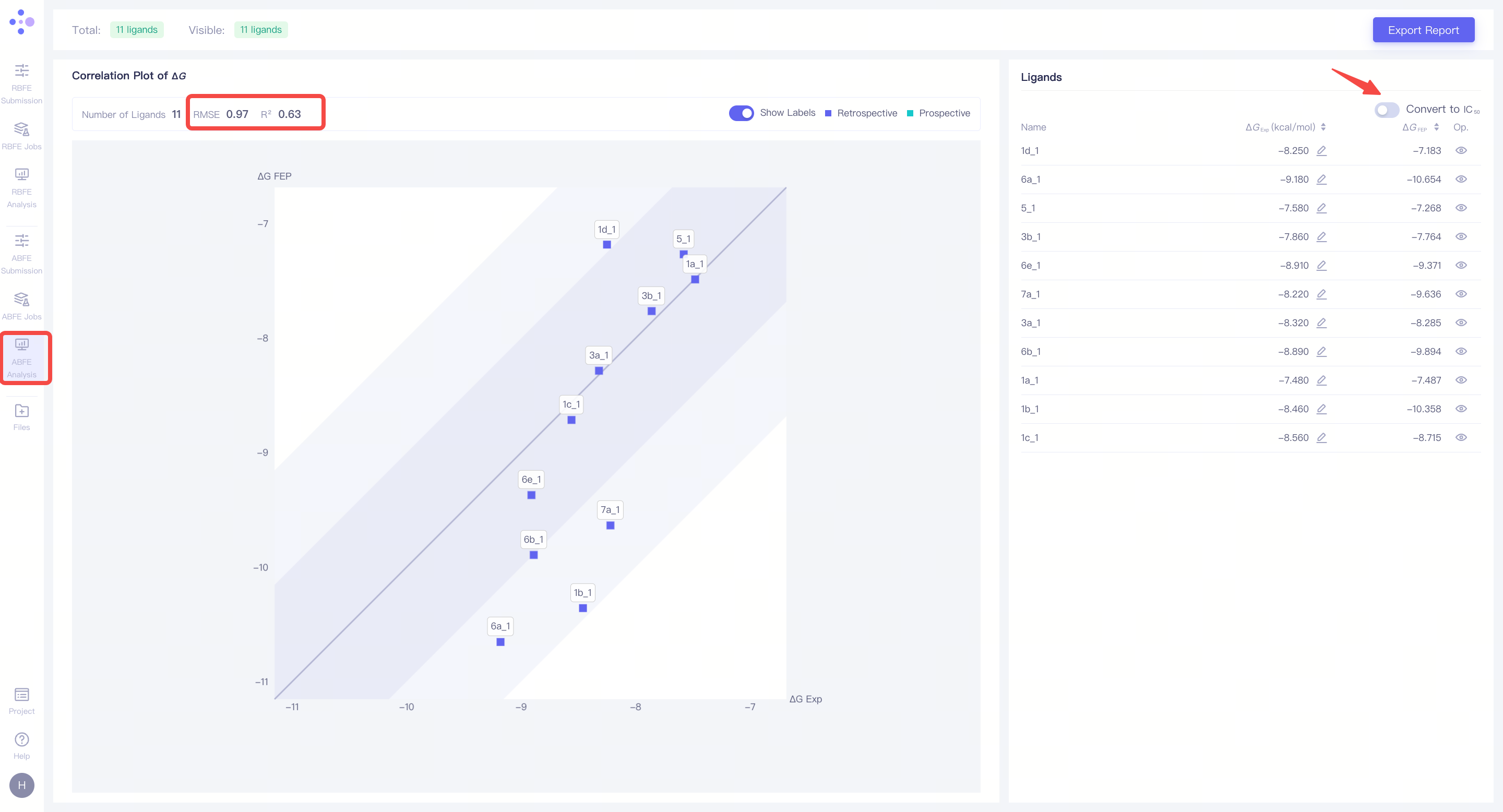
| 分子整体计算结果相关性分析:��将左视图切换到 "Correlation Plot of ΔG",右视图切换到 "Ligands",可以对每个分子的 FEP 计算结果进行全面分析。左上角将显示整体 FEP 计算的 相关性系数 (R²) 和 均方根误差 (RMSE)。当分子的活性实验值跨度较大时,建议参考 R² 值,R² 越接近 1,说明计算结果越准确,通常 R² > 0.4 可以接受。当分子活性值较接近时,建议参考 RMSE 值,RMSE 越小 说明计算结果越好,通常 RMSE < 1.4 kcal/mol 是可接受的范围。 |
|
| 确认FEP模型的可靠性:通过上述热力学循环一致性、ΔΔG 相关性和 ΔG 相关性三个维度的分析,如果所有指标均符合预期,可以认为本次 FEP 计算结果是可靠的,构建的 FEP 模型已达到生产级别,可以用于 未测定活性分子 的活性预测。 |
4. 使用Uni-FEP进行ABFE计算 - 操作清单
| 小贴士:不熟悉ABFE计算的读者可以先跳过本章 本章节旨在给熟悉ABFE计算的用户一个清晰简要的使用指南,以确认每一个重要步骤都完成操作。 如果您首次接触ABFE计算,可以先阅读第五章节,通过案例和可视化界面截图了解如何使用Hermite Uni-FEP完成一次完整的ABFE计算和分析工作。再以本章节的内容作为实操过程的指引。 |
4.1 ABFE 数据收集与基本检查
收集并检查蛋白结构选择高质量的晶体复合物结构作为起始蛋白质模型,确保结合口袋附近的氨基酸残基清晰解析,蛋白质链无断裂。
收集并检查分子结构和性质确保部分分子具备可靠的实验数据,且数据来源统一,覆盖较大的结合亲和力范围。
4.2 ABFE 体系准备与任务提交
上传蛋白并进行准备上传蛋白结构并进行蛋白预处理流程,保留必要的氨基酸链、水分子和其他辅助结构,设定氨基酸的质子态。
蛋白预处理后,系统会进行自动检查。若检查未通过,根据反馈修正结构。
若结合位点中存在关键的可质子化氨基酸残基(如Asp、Glu、His等),请仔细确认并根据结合模式手动调整其质子化状态,以确保计算的准确性。
如蛋白系统中包含对功能或结合模式具有重要影响的辅因子(如NAD、FAD、金属离子等),请将其一并上传并确认其结构与状态的合理性。
若目标蛋白为膜蛋白,请在预处理后搭建合适的磷脂双分子层。
上传配体分子并进行分子对接上传配体分子并进行自动检查。若检查未通过,根据反馈结果修正结构。
对于未确定结合结构的配体,开展分子对接。并通过相互作用模式选择最合理的结合模式作为后续进行自由能计算的结构。
如配体分子具备实验测定的结合亲和力数据,需导入该数据。
设定计算参数并提交计算任务进入提交前的确认页面,核实所使用的蛋白质和待计算的分子是否符合预期。
检查并确认 ABFE 计算的模拟参数,确保设置正确。
确认即将消耗的 FEP pairs 数,确保账户余额足以完成本次计算。
点击提交,ABFE 计算任务将自动分配至计算集群。
4.3 ABFE 任务监控与结果分析
监测任务进程和状态在 ABFE 任务界面,监控每一个分子的模拟进程和状态。
若某个分子的模拟失败,需检查并修改其结合姿态和模拟参数,考虑重新提交。
分析所有分子的计算结果通过相关性图,查看 ABFE 计算所得的结合自由能与实验数据的相关性,评估 ABFE 计算模型的可信度。
基于 ABFE 计算结果,对未测定活性的分子进行计算评估。
5. 使用Uni-FEP进行ABFE计算 - 以Thrombin体系为例
Thrombin 蛋白是一种在血液凝固过程中起关键作用的酶。在凝血级联反应中,它通过将纤维蛋白原转化为纤维蛋白,从而形成血凝块。抑制或调节 Thrombin 的活性可以直接影响血液凝固过程。因此,开发 Thrombin 蛋白的小分子抑制剂对于治疗血栓类疾病和抗凝药物至关重要。
在本章节中,我们将基于已报导的药物研发项目,以Thrombin 蛋白为目标,展示如何利用 Uni-FEP 进行自由能微扰计算的体系构建与验证。本案例来自 Bernhard Baum 等人2009年于 Journal of Molecular Biology 发表的研究[1],该工作深入探讨了药物分子在 Thrombin S1 特异性口袋中的结合机制,结合 X-ray晶体结构解析 等多种实验手段,揭示了Asp189和Tyr228等关键残基形成的相互作用在结合自由能中的关键贡献。
| [1] Baum B, Mohamed M, Zayed M, Gerlach C, Heine A, Hangauer D, Klebe G. More than a simple lipophilic contact: a detailed thermodynamic analysis of nonbasic residues in the S1 pocket of thrombin. Journal of molecular biology. 2009 Jul 3;390(1):56-69. |
5.1 ABFE 数据收集与基本检查
本教程所使用示例数据:
thrombin_core_protein.pdb
thrombin_core_ligands.sdf
5.1.1 收集并检查蛋白结构
我们选取了 2ZFF 晶体结构作为参考模型,该结构解析了 Thrombin 蛋白与未取代苯基抑制剂 5 的复合物,分辨率高达 1.47 Å,提供了清晰且高质量的结合口袋构象,尤其是 S1 特异性口袋中关键残基(如 Asp189 和 Tyr228)的空间位置。这为 FEP 计算提供了可靠的起始蛋白结构,并确保结合口袋内的相互作用模式具有较高可信度。
图:Thrombin 蛋白与抑制剂的复合物晶体结构(PDB ID:2ZFF)
5.1.2 收集并检查分子结构和性质
我们选择了结构相似但取代基不同的一系列抑制剂,包括 1a, 1b, 1c, 1d, 3a, 3b, 5, 6a, 6b, 6e 和 7a。这些分子具有高度一致的核心骨架,而在 S1 口袋内的苯基取代基表现出系统性的变化,例如氯、氟、溴和甲基等取代基,能够分别反映疏水性、体积及特殊相互作用对结合驱动力的贡献。此外,这些分子覆盖了实验测定的较宽结合自由能范围,为 FEP 模型的验证提供了完整且统一的热力学数据支持。
图:Thrombin 蛋白抑制剂的化合物结构和结合自由能
5.2 ABFE 体系准备与任务提交
5.2.1 创建FEP项目并进入项目页面
| 操作描述 | 界面截图 |
| 在 Hermite 项目列表 页面中,点击右上角的 "+ New" 按钮,输入项目名称,选择项目类型为 "FEP",然后点击 "Apply",即可创建一个 FEP 项目并进入项目页面。 |
|
| 左侧菜单栏切换到"ABFE Submissioon",进入ABFE任务提交界面。 |
|
5.2.2 上传蛋白并进行准备和检查
蛋白质准备是 FEP 计算的重要步骤,确保蛋白质结构的合法性、准确性和稳定性。本小节将分步骤指导您如何完成蛋白质的上传、结构检查、质子化调整等操作。
| 操作描述 | 界面截图 | ||||
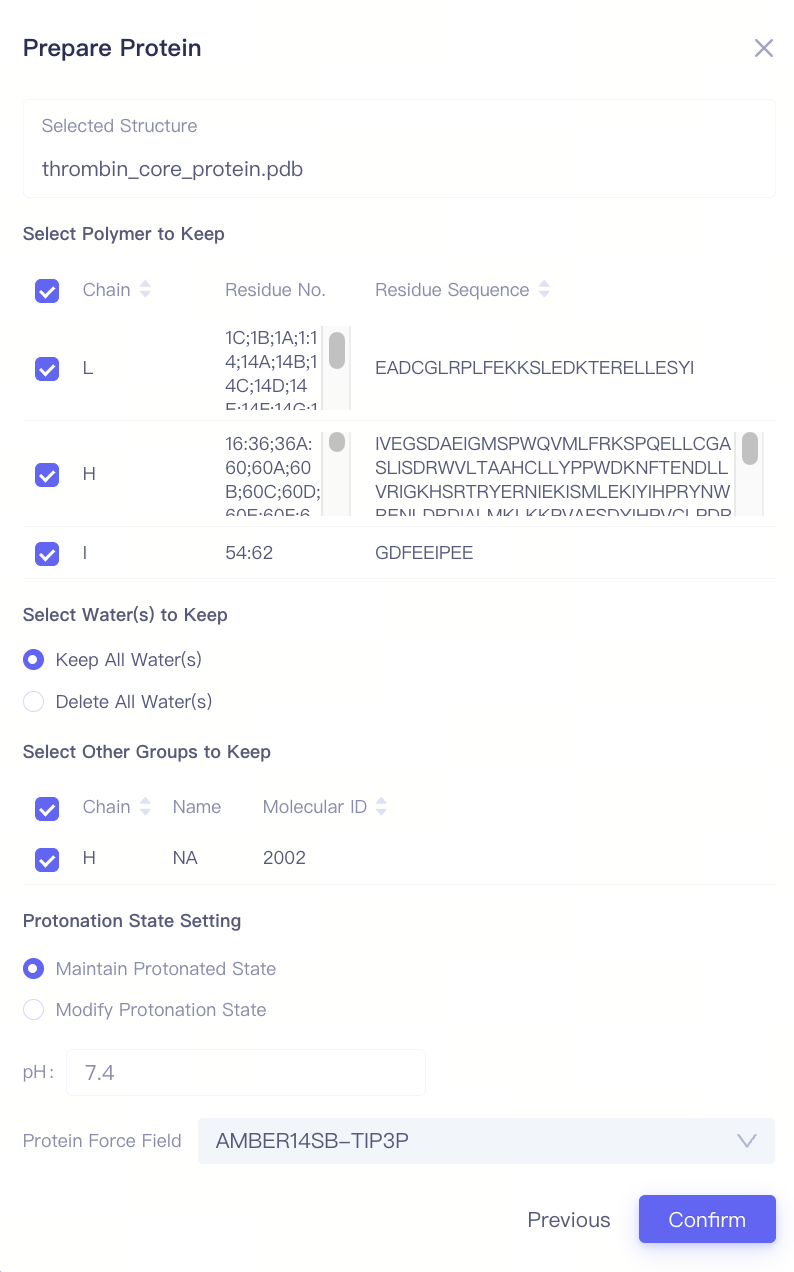
| 上传蛋白结构文件并进行准备:在页面左上角蛋白窗格,点击 "Upload a Protein" 按钮,选择要上传的蛋白结构文件(例如 thrombin_core_protein.pdb),然后点击 "Next" 进入蛋白准备界面。 选择保留的氨基酸链:在 "Select Polymer to Keep" 选项中,选择需要保留的氨基酸链。一般来说,应保留与配体结合位点相关的氨基酸链,以确保后续计算的准确性。此处保留所有的氨基酸链。 选择保留的水分子:在 "Select Water(s) to Keep" 选项中,选择是否保留晶体结构中的水分子。如果上传结构中存在:(1)深埋在蛋白内部且对整体结构稳定性重要的水分子; (2)参与蛋白-配体相互作用的水分子,则建议选择"Keep All Water(s)",否则可以选择"Delete All Water(s)"删除水分子,以减少计算负担。此处选择"Keep All Water(s)"。 选择保留的其他分子或化学基团:在 "Select Other Groups to Keep" 选项中,检查是否有需要保留的其他分子或化学基团(例如辅助因子、共价修饰基团等)。此处保留H链中的NA。 调整氨基酸侧链质子化状态:在 "Protonation State Setting" 中,选择质子化状态设置。如果您已经确认氨基酸的质子化情况,使用 "Maintain Protonated State",以保留晶体结构中氨基酸侧链的质子化状态。否则,您可以使用"Modify Protonation State"让程序根据您设置的pH值自动调整氨基酸的质子化状态。此处蛋白已经过处理,具有合适的质子化状态,因此选择 "Maintain Protonated State"。 选择蛋白质力场:在 "Protein Force Field" 中,选择合适的力场(例如 "AMBER14SB-TIP3P"),以确保在分子动力学模拟中蛋白的能量计算准确可靠。此处默认选择"AMBER14SB-TIP3P"。 确认蛋白结构设置:完成上述设置后,点击 "Confirm" 按钮,系统将开始蛋白结构的预处理和合法性检查。通常,这一过程会持续约1分钟。 |
| ||||
| 检查蛋白准备状态:蛋白准备完成后,系统将显示检查结果状态,包括 "Valid"(有效)、"Warning"(警告)或 "Error"(错误)。您可以根据提示进行修正或进一步检查。 |
| ||||
| 手动调整质子化状态:如果检查结果中存在质子化状态的警告或蛋白配体结合位点附近有重要的可质子化氨基酸,您可以点击 "Modify Protonation States" 选项,对质子化状态进行手动检查和调整。 |
| ||||
| 添加非标准组分:如果蛋白质结合位点附近存在非标准组分(例如辅助因子或金属离子),请在 "Non-standard Components List" 中上传相关的配体结构或构建好的力场参数文件,以确保模拟的准确性。 |
| ||||
| 构建膜环境:如果目标蛋白是膜蛋白,请点击 "Build Membrane" 进入膜构建界面。在该界面中,您可以调整膜的厚度、溶剂填充层以及平衡时间等参数,确保蛋白正确嵌入膜中。 |
| ||||
| 折叠蛋白窗格:完成上述步骤后,可以点击折叠按钮来隐藏蛋白设置的相关组件以节约屏幕空间。 |
|
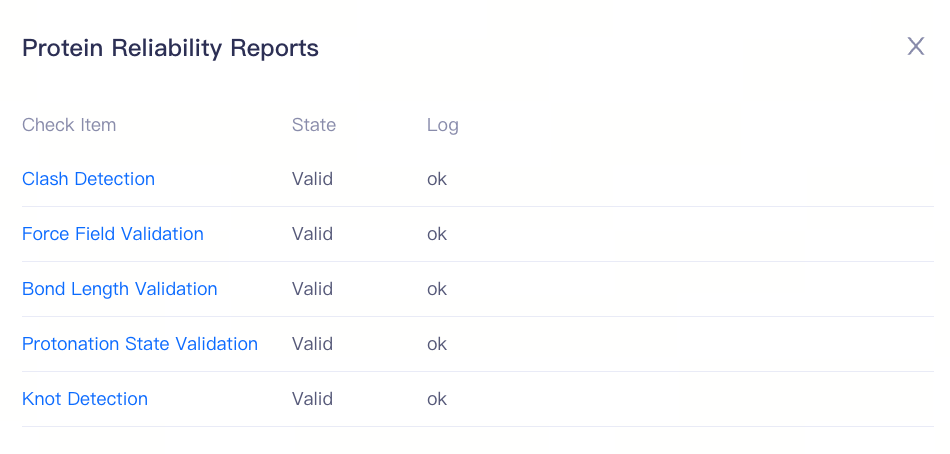
| 小贴士:蛋白合法性检查的项目及可能的状态 碰撞检查(Clash Detection)[Valid/Error]:检测蛋白结构中是否存在原子间的不合理碰撞,常见问题包括原子重叠、非物理性接近、可替换残基异常、金属离子未配位等,这些问题可能导致模拟不稳定或能量极小化失败。 力场参数检查(Force Field Validation)[Valid/Error]:确保蛋白质结构中的所有组分均具备完整且合适的力场参数,以便在后续分子动力学模拟中准确计算相互作用能量。 键长检查(Bond Length Validation)[Valid/Error]:验证蛋白质中所有化学键的长度是否在合理范围内,避免异常的拉伸或压缩,从��而确保分子动力学模拟的稳定性。 质子化状态检查(Protonation State Validation)[Valid/Warning]:确保蛋白质中的氨基酸残基和配体的质子化状态在指定 pH 条件下是合理的,尤其是活性位点残基(如组氨酸、谷氨酸、天冬氨酸等),避免电荷分布错误影响结合自由能计算。 交叉缠结检查(Knot Detection)[Valid/Error]:检测蛋白质主链是否存在异常的交叉缠结结构(knot),即主链在空间中互相穿插缠绕的情况。这种异常通常源自实验解析误差或建模失误,可能导致几何优化、能量极小化、分子动力学模拟过程中的严重不稳定,甚至引发模拟失败。 |
5.2.3 上传配体分子并进行处理
配体准备是进行 FEP 计算的重要前置步骤,确保配体分子结构的合法性、准确性和构象合理。本小节将分步骤指导您如何进行配体的上传、检查、对接、叠合和实验数据的导入。
| 操作描述 | 界面截图 |
| 上传配体分子结构文件:在页面左上角的 配体窗格 中,点击 "Upload Ligand(s)" 按钮,选择要上传的配体分子结构文件(例如 thrombin_core_ligands.sdf)。系统会自动进行配体的合法性检查。如果上传的 .sdf 文件中包含多个配体,系统将自动将其拆分为多个独立的配体条目。 |
|
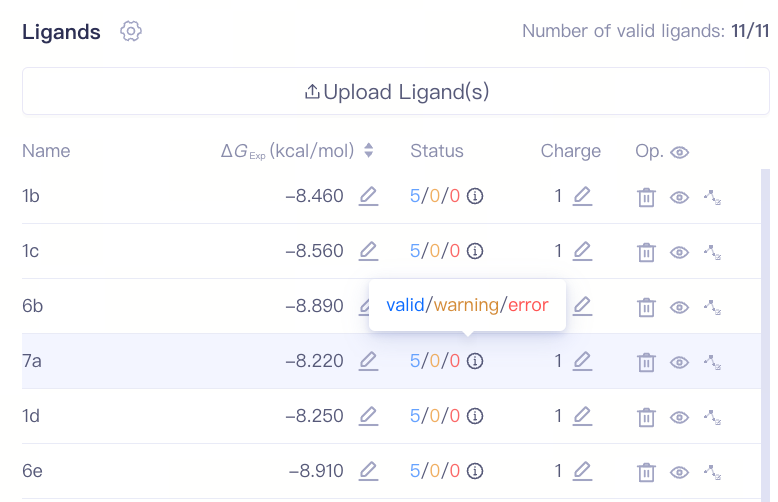
| 检查配体合法性:配体检查完成后,系统将显示检查结果状态,包括 "Valid"(有效)、"Warning"(警告)或 "Error"(错误)。您可以根据提示进行修正或进一步检查。Charge列展示了该分子的总电荷数,您可检查所上传分子的质子化状态是否正确,如果不正确,您可通过右侧的编辑按钮修改该分子的总电荷数。 |
|
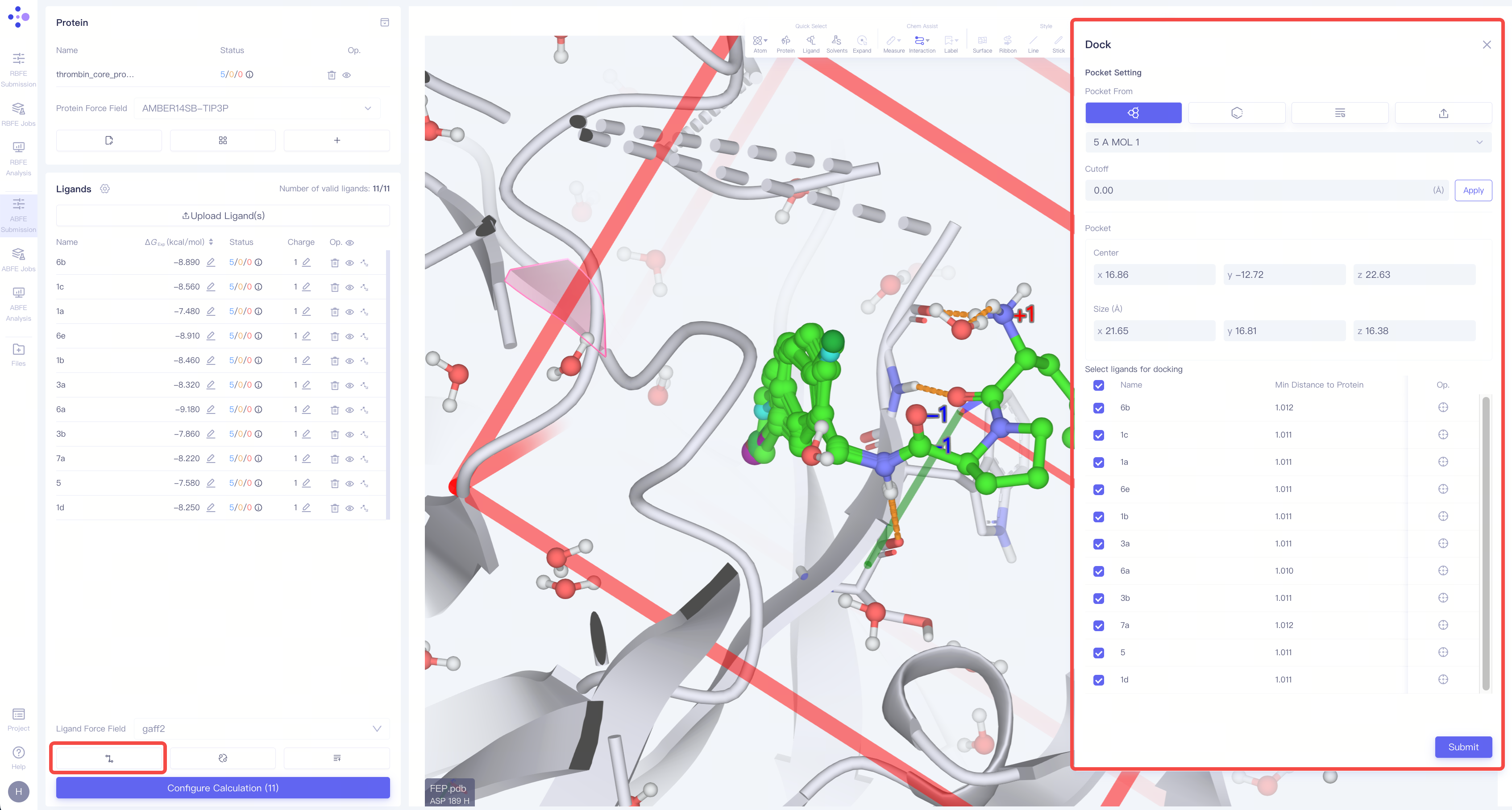
| 配体分子对接:在 3D 窗格 中,检查上传的配体是否具有合适的构象。如果构象不合适,请点击左下角的 "Dock Ligands" 进行分子对接操作。 指定对接盒子:Pocket Setting处指定口袋位置及大小; 选择需对接的分子:Select ligands for docking处选择需要对接的分子。 点击"Submit"提交分子对接计算。 计算完成后,可通过配体列表中的"Slect pose"查看及选择对接姿态。 |
|
| 上传实验活性数据:如果配体分子具有已测定的实验活性数据(例如 IC₅₀ 或 Ki 值),可以直接在系统中编辑相应分子的实验值,或点击右下角的 "Upload Affinity Data",通过模板文件一键批量上传实验数据,确保实验值与配体分子正确关联。 |
|

| 小贴士:配体分子合法性检查的项目及可能的状态 力场参数检查(Force Field Parameter Validation)[Valid/Error]:确保配体的每个原子类型都被正确识别,并分配了合适的力场参数,包括键长、键角、二面角以及电荷分布,以保证能量计算的准确性。 总电荷检查(Total Charge Validation)[Valid/Warning]:检查配体的实际总电荷是否与通过化学结构推导出的理论电荷相一致。在质子化状态不明确或存在建模错误时,配体的总电荷可能偏离预期,从而影响电荷相关的能量计算过程。 配体类型检查(Ligand Type Detection)[Valid/Error]:根据配体的原子数和组成判断其是否为合理的小分子药物候选物。若原子数小于5,通常被判定为金属离子、缓冲离子或溶剂分子等非药物分子类型,不能作为配体参与结合自由能计算。 配体-蛋白距离检查(Protein-Ligand Distance Detection)[Valid/Warning]:检查配体与蛋白质之间的空间距离,确保配体处于合理的结合位点,避免非物理性接近或过远距离,这可能导致结合自由能预测偏差。 |
5.2.4 设定计算参数并提交计算任务
| 操作描述 | 界面截图 |
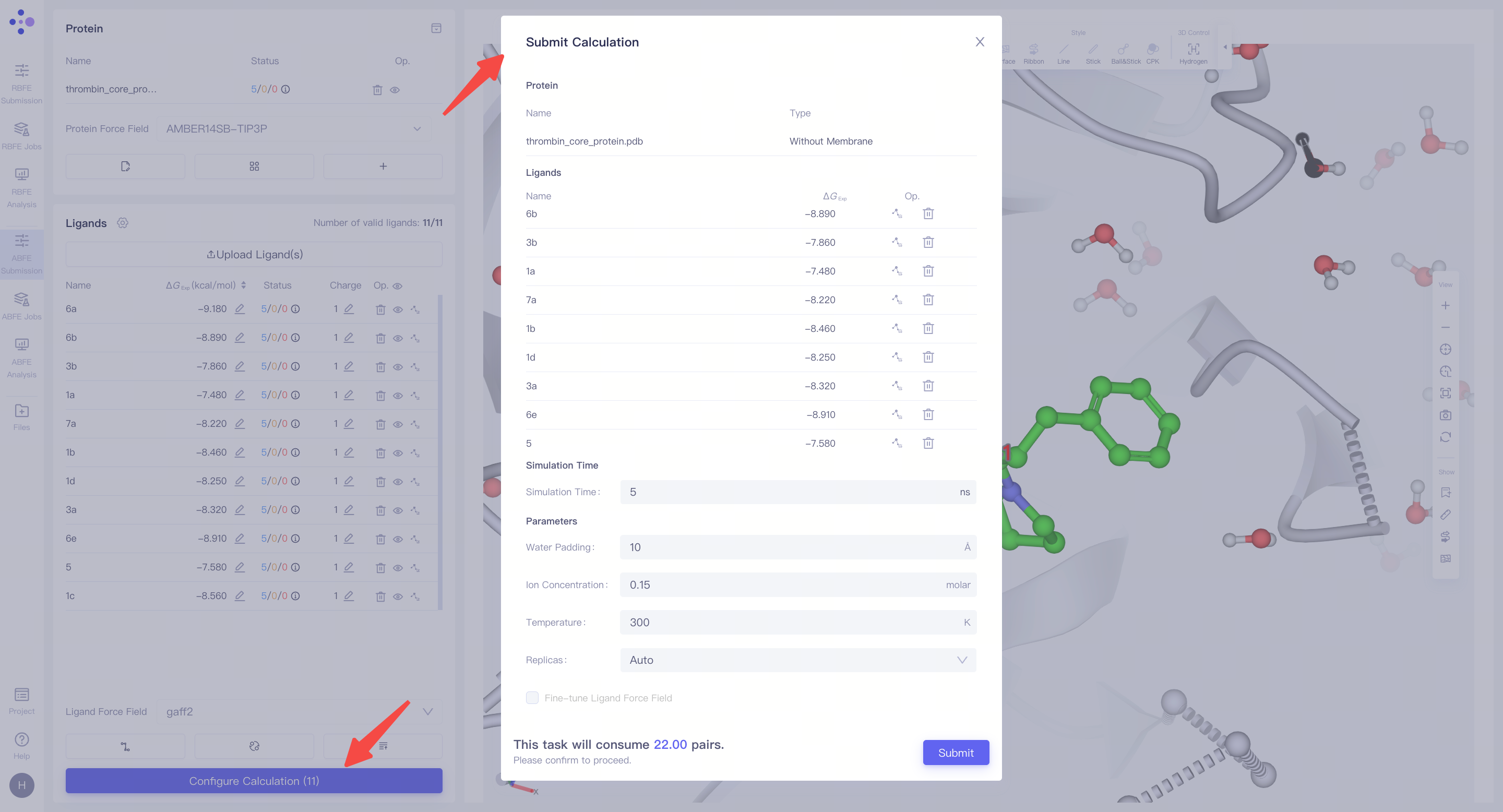
| 进入计算参数配置界面:当所有前置任务完成后,右下角的 "Configure Calculation" 按钮将变为可点击状态。点击该按钮进入 FEP 计算参数配置 界面,开始进行任务的详细设置。 确认��膜蛋白体系标识:如果目标体系是 膜蛋白,请确保 "Type" 列中已标记为 "with Membrane",以保证模拟条件正确匹配膜环境。 设置模拟时长(Simulation Time):对于常见体系,建议将模拟时长设置为 5 ns,以确保计算结果的稳定性和准确性。 配置模拟参数:设置添加水分子的层厚(Water Padding),以确保体系边界不会影响模拟结果;设置离子浓度(Ion Concentration),离子为 Na⁺ 和 Cl⁻,用于中和体系电荷并模拟生理环境;设置模拟温度(Temperature),通常为 300 K,以匹配常规实验条件。 确认计算费用:完成所有参数设置后,系统将自动统计本批次 FEP 计算任务的预计费用。请仔细核对所有设置和计算资源信息,确保无误。 提交计算任务:点击 "Submit" 按钮,所有 FEP 计算任务将被提交到服务器,开始正式计算。 |
|
5.3 ABFE 任务监控与结果分析
5.3.1 监测任务进程和状态
| 操作描述 | 界面截图 |
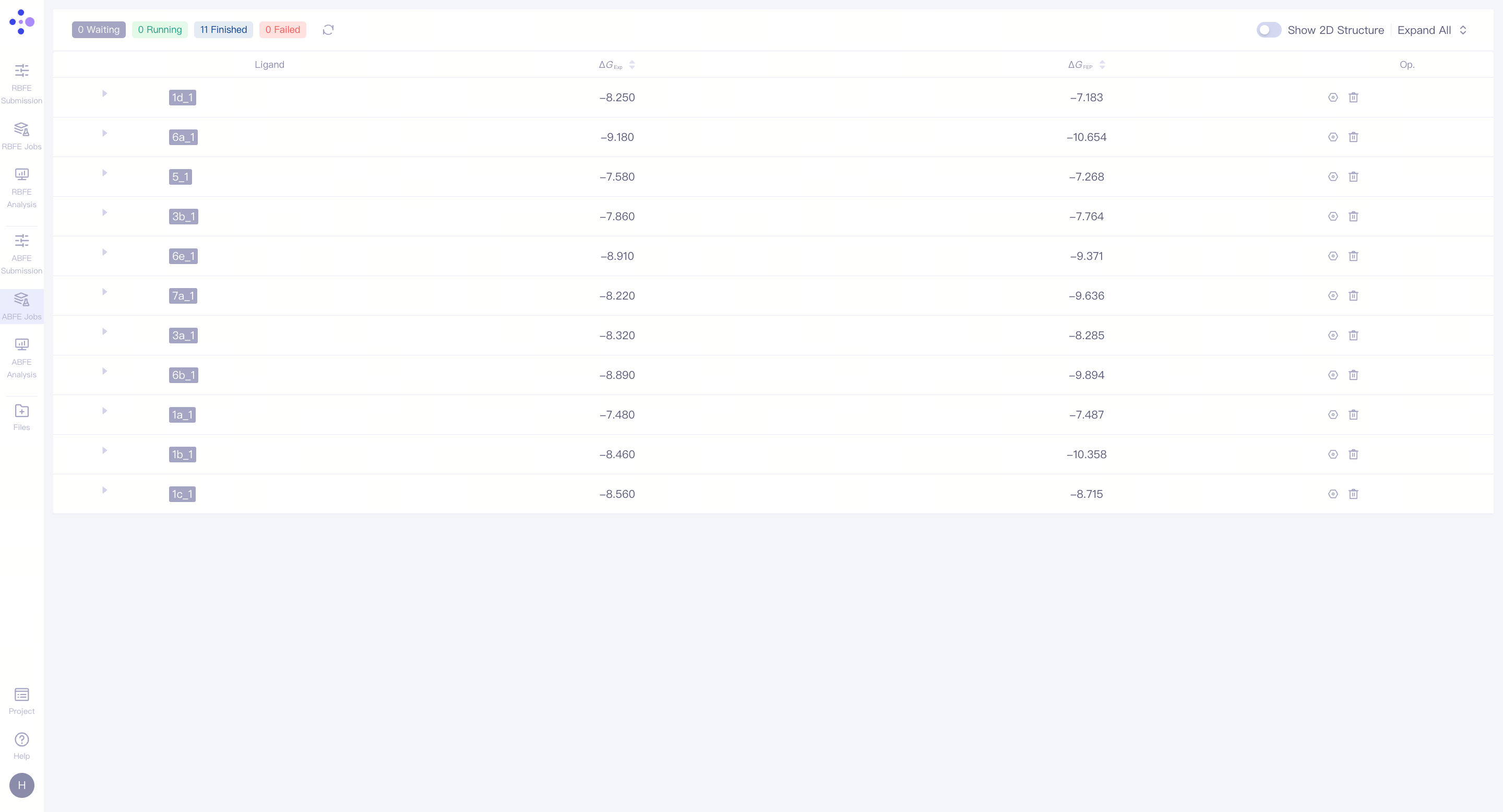
| 进入 FEP 任务管理界面:在左侧侧边栏中,点击 "ABFE Jobs",进入 FEP 任务管理 界面 页面左上角:展示了所有任务的 状态统计,包括已完成、进行中、失败等不同状态的任务数量。 页面右上角:选项按钮,允许用户切换分子对的 2D 结构图片展示,以及是否显示每个分子对的模拟信息,以便根据需求自定义界面视图。 |
|
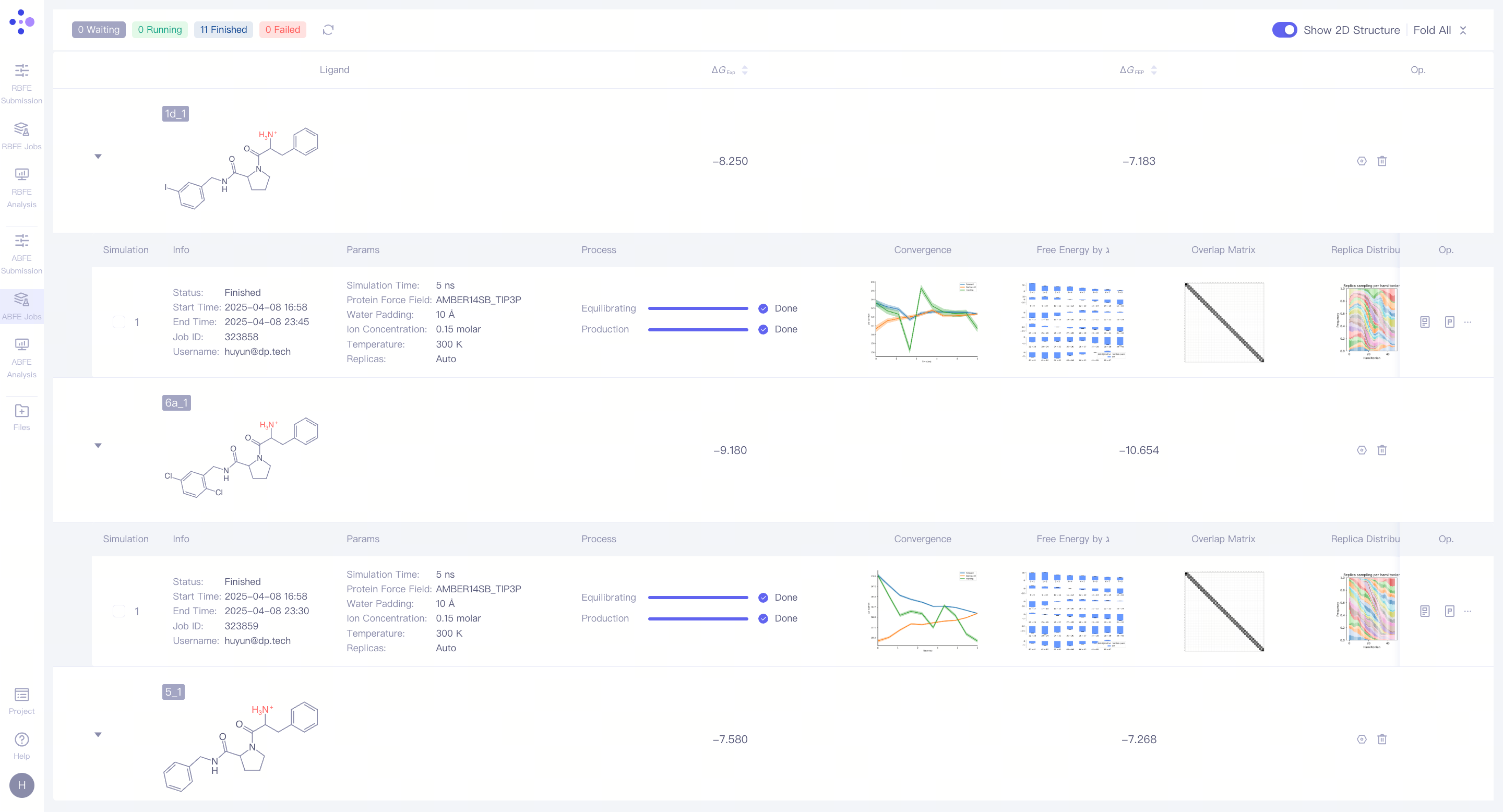
| 单任务管理:ABFE 任务管理页面展示每个分子计算任务的信息及结果。 展开任务详情:点击每个分子对左侧的 三角箭头,可以展开显示该分子对的详细模拟信息。展开分子对的模拟信息后,您可以查看包括基本信息、模拟参数、任务进度、结果图示,还有相对应的操作按钮。 监控任务进度:任务有等待(Waiting)、平衡(Equilibrating)、生产(Production)三种任务状态。对处于模拟阶段的 任务,可以观测其运行进度和预期结束时间。 |
|
5.3.2 分析单个分子的计算结果
单个分子的 FEP 计算完成后,可以通过一系列可视化分析工具对模拟结果进行详细解析,评估计算的准确性和可靠性。本小节将指导您如何解读和分析单个分子对的计算结果。
| 操作描述 | 界面截图 |
| 查看计算结果:当单个模拟任务成功完成后,左上角会显示 任务完成 状态,同时,该分子的 ΔG_FEP 数值将自动计算并展示在界面上,作为分子绝对结合自由能的预测结果。 |
|
| 分析模拟收敛性:展开任务详情,可以观察本次 FEP 计算的 模拟收敛性,系统提供以下四种关键分析图表: 收敛性分析:收敛性分析展示了前向与后向自由能结果之间的差异,这种差异反映了滞后效应的程度。在理想情况下,前后向结果的差异应非常小,表明体系已经充分收敛,采样偏差可忽略不计。如果差异较大,通常意味着状态重叠不足或采样不充分。此外,平滑且稳定的移动平均值暗示自由能数值已趋于收敛,而持续变化的移动平均值则说明体系尚未完全收敛,仍在演化中。 自由能 λ 分解:自由能λ分解反映了体系在不同λ状态下的自由能变化情况。如果ΔΔG在λ区间内呈现平滑变化,说明体系在炼金转换过程中的过渡较为稳定,系统表现良好。若在某些λ值处出现突兀的跃变,可能暗示该阶段存在关键转换点,或采样不足,亦或是体系存在不稳定性。 相空间重叠:相空间重叠矩阵展示了不同λ状态之间的构象采样重叠度。每个单元格的数值代表两个相邻λ窗口之间的构象空间共享程度,数值越高(通常建议大于 0.15),表示相邻状态之间的过渡更平滑,计算结果也更可靠。如果某些区域的重叠度较低,可能表明状态之间存在显著的能垒或采样不足。 副本分布:副本分布展示了不同λ区间的采样覆盖情况。均匀分布意味着所有λ状态都得到了充分的采样,整个系统在所有窗口中的采样均匀且稳定。如果分布不均匀,可能暗示某些λ区间存在采样瓶颈或能量壁垒,影响最终自由能的准确性。 更详细的分析结果可以通过点击 "Report" 下载完整的报告文件进行深入解析。 |
|
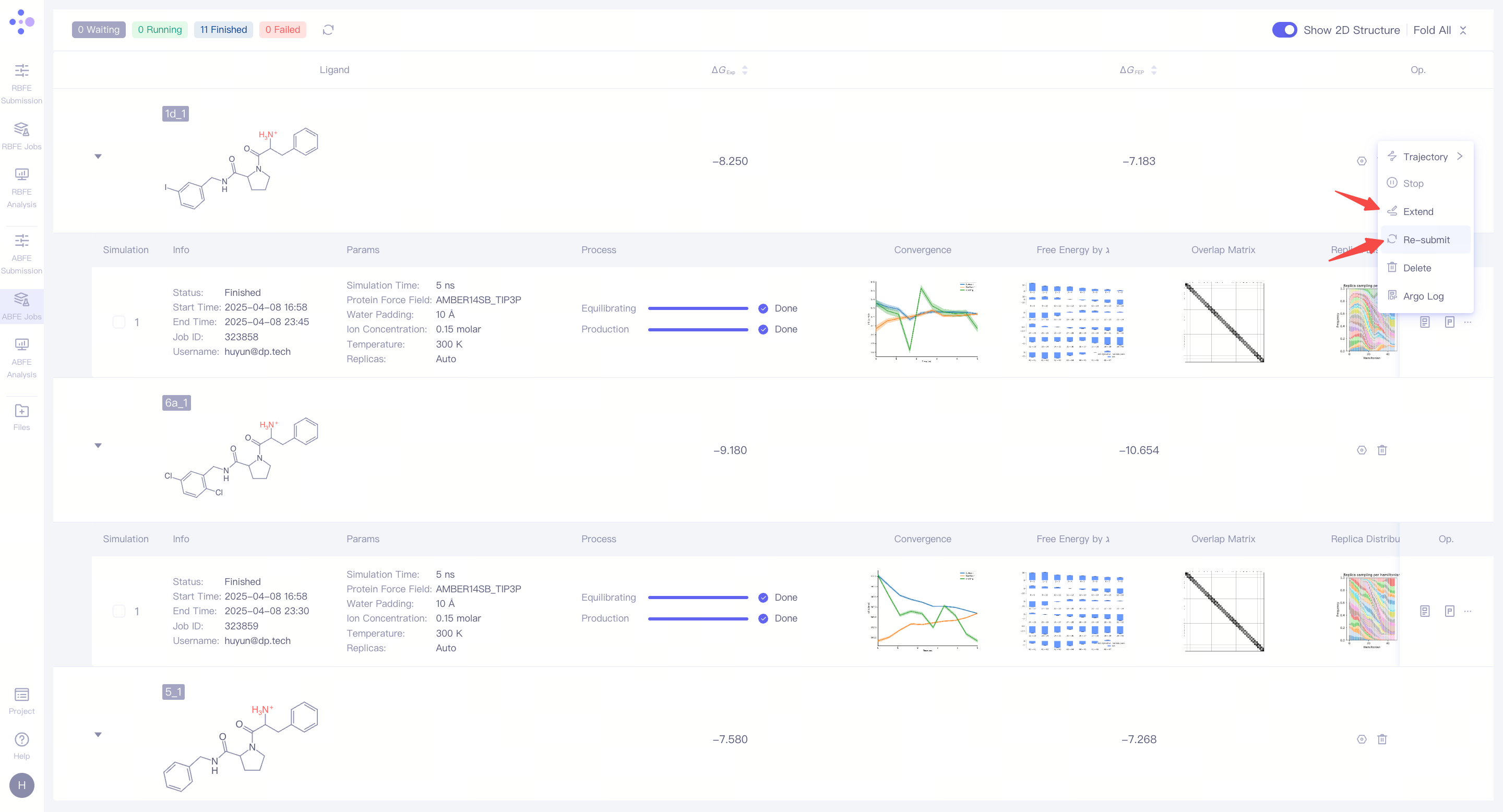
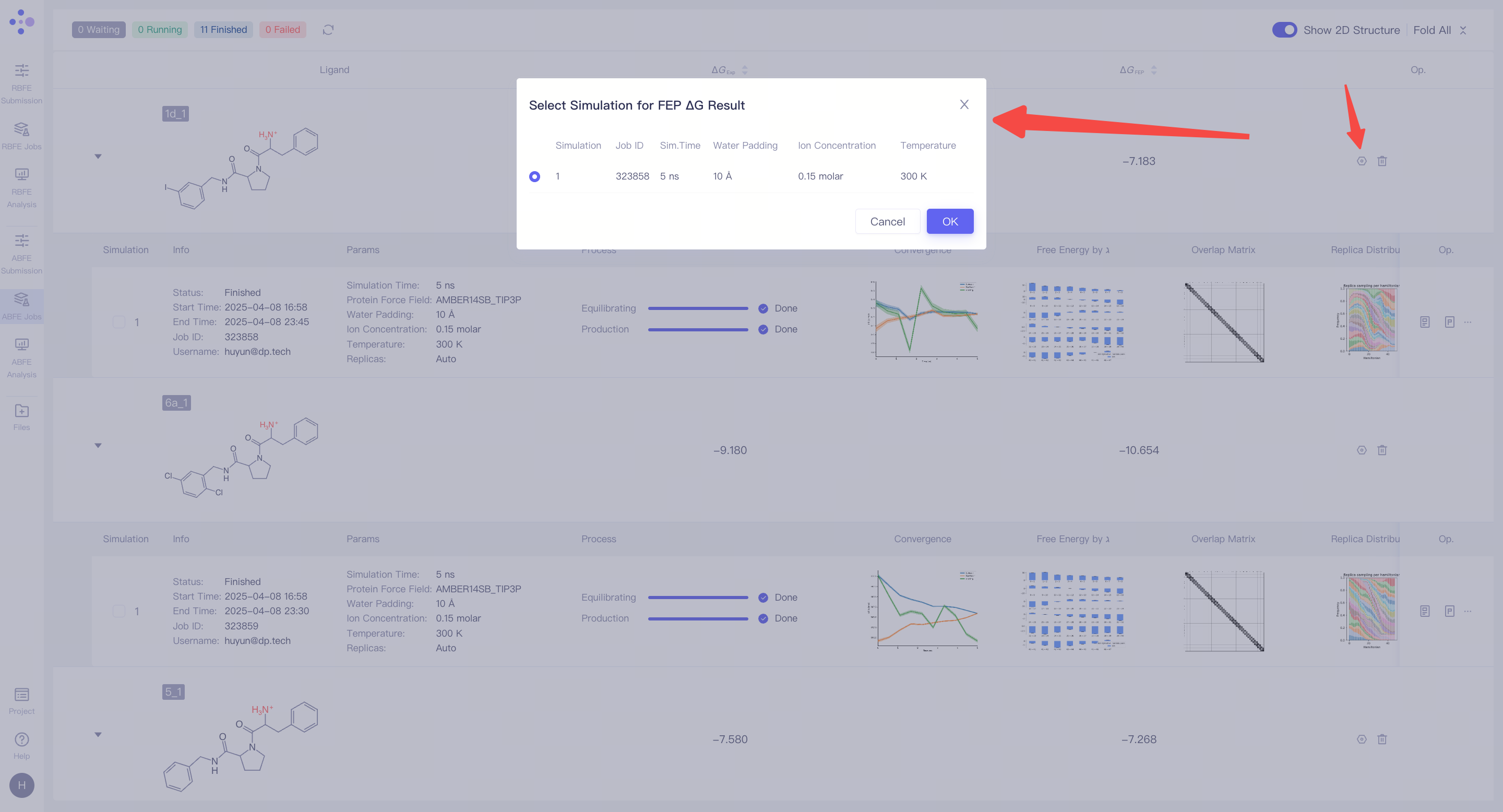
| 处理未收敛或不满意的结果:如果发现模拟结果未收敛或结果不理想,可以点击 "Extend" 按钮,延长模拟时间,增加采样量以提高结果的稳定性。如果需要重新进行计算,可以点击 "Re-submit" 按钮,重新启动该分子的模拟任务。在重新提交的界面可以修改所选择的结合姿态。当一个分子对有多个模拟结果时,可以选择其中某一次特定的模拟作为最终的 ΔG_FEP 计算来源(系统默认选择最近一次的模拟结果)。 |
|
5.3.3 汇总所有分子的计算结果
| 操作描述 | 界面截图 |
| 进入分析界面:当所有分子对的计算任务完成,并确认每对分子对的模拟结果已收敛后,点击左侧侧边栏的 "ABFE Analysis" 进入整体结果分析界面。在该界面中,用户可以基于ΔG 相关性进行分析。 | |
| 分子整体计算结果相关性分析:在"Correlation Plot of ΔG"处可以对每个分子的 FEP 计算结果进行全面分析。左上角将显示整体 FEP 计算的 相关性系数 (R²) 和 均方根误差 (RMSE)。当��分子的活性实验值跨度较大时,建议参考 R² 值,R² 越接近 1,说明计算结果越准确,通常 R² > 0.4 可以接受。当分子活性值较接近时,建议参考 RMSE 值,RMSE 越小 说明计算结果越好,通常 RMSE < 1.4 kcal/mol 是可接受的范围。 |
|
| 确认FEP模型的可靠性:通过 ΔG 相关性分析,如果指标均符合预期,可以认为本次 FEP 计算结果是可靠的,构建的 FEP 模型已达到生产级别,可以用于 未测定活性分子 的活性预测。 |
6. 进阶技巧
当计算两个分子差异较大时,为什么要构建过渡分子?
当两个分子在骨架上或官能团类型上差异明显时,直接进行自由能差的计算往往导致映射关系复杂、采样不充分,可能出现结果波动过大或难以收敛。为了让体系逐步过渡,建议在 Uni-FEP 中手动或自动生成若干“过渡分子”,将大改动拆分为多个小改动。例如, A 分子与 B 分子同时在两个官能团(如 R1 和 R2)上发生了改变,直接将 A 变为 B 可能造成体系瞬间的大幅度变化,导致映射关系复杂、采样不充分。此时可以构建一个或多个“过渡分子”:例如先只将 A 的 R1 改成 B 的 R1(而保持 R2 不变),得到一个中间分子 C;然后再在 C 的基础上,把 R2 改成 B 的 R2。这样分两步就能完成 A→B 的转换,比“一步到位”更易收敛。在 Uni-FEP 中,您可以创建并上传这些中间分子,让软件分段完成自由能计算,显著提高准确度并降低计算风险。

当候选分子与参考分子叠合(Alignment)效果不好时,如何利用其他相似分子做参考?
如果两个分子无法直接通过叠合模块(Ligand Alignment)得到稳定且可比的初始构象,可以选择与它们骨架或官能团最相似的“中间分子”作为叠合参考,将候选分子和参考分子分别与该中间分子进行对齐。例如,A 分子是单环,B 分子是多环,直接叠合时常出现错位或旋转不匹配的问题。若体系中有一个与二者都更接近的“中间分子”C(例如骨架接近 B,但外围取代基类似 A),可先用 Uni-FEP 对 A 与 C 进行精细叠合,随后再让 B 与 C 叠合,并在映射设置时指定 C 为共同参考。这样往往能得到更可靠的初始构象,避免后续 FEP 计算出现大规模重排或不收敛。
检查分子映射(Mapping)时,需要重点关注哪些原子或基团?
在 FEP 计算中,映射时若将不相容的基团对应在一起,经常会引发能量骤升或“原子交叉”。(1) 如果遇到芳香环与脂肪环的转换,要注意是否把苯环的平面结构硬塞到环己烷的非平面构象上;若发现自动映射让环系相互交错,可以在软件的“编辑映射”功能里手动指定环上碳原子的对应关系。(2) 对于线性氰基(-CN)这类基团,若目标分子是 -NO₂、-NH₂ 之类物性相差极大的官能团,最好在正式计算前构建一步或多步过渡分子,避免一步到位的巨大电子结构变化。另外,对于含手性中心的分子,要确认 R/S 构型不会被自动映射所颠倒;一旦发现立体翻转,可以通过手动修改映射并在可视化界面检验其合理性。
如何根据蛋白-配体的结合姿态来决定关键残基的质子化状态?
蛋白中可质子化的氨基酸(如组氨酸、赖氨酸、谷氨酸等)与配体之间的局部环境往往会改变它们在生理条件下的酸碱行为。(1) 如果配体上带有一个带正电的基团,但与组氨酸侧链只是空间上相邻而无明显氢键,可考虑该组氨酸是否仍倾向于质子化,以便与配体形成静电吸引;相反,如果配体的羧酸基团与谷氨酸侧链处在 3 Å 以内并形成稳定氢键或盐桥,则谷氨酸更可能处于去质子化态。(2) 在实际操作中,可以结合软件的 pKa 预测、距离分析和晶体结构信息来做判断;如果自动分配与已知结合模式不符,要手动修正该残基的质子化,并在后续所有分子的计算中保持统一的蛋白版本。
为什么要基于热力学循环来删减不必要的微扰边?
当要比对多条分子间的相对自由能差时,往往会形成一个包含 A、B、C、D 等多个节点和相互连接边的网络。如果某条“边”(例如 A→D)涉及同时改变多个官能团或存在映射不良,可能会产生很高的收敛难度或波动。(1) 在这种情况下,可以先通过 A→B、B→C、C→D 等较为平滑的过渡,让每一步的改动合理且易收敛,然后再利用热力学循环推导出 A→D 的结果。(2) 若经过评估发现某些边始终是误差源头,不如直接将其从计算网络中删去,保留循环闭合度更高的路径,既能减少计算量也能提升整体预测的准确度。
进行分子叠合(Ligands Align)的时候,如果把目标分子叠合到参考分子的效果不理想,怎么办?
方法一:在选择待叠合分子的界面中,可通过操作(Op.)列中的编辑按钮,手动调整原子映射(Atom Mapping),确保目标分子与参考分子的公共子结构精确对应,以优化叠合效果。
方法二:若目标分子与参考分子结构差异较大,建议先选择与参考分子结构相似的分子进行叠合。然后,将已成功叠合且与目标分子结构差异较小的分子作为新的参考分子,再进行叠合操作,以提高叠合的准确性和稳定性。
骨架跃迁(Core Hopping)体系中常见的原子映射(Atom Mapping)连接建议。
将五元环变为六元环的过程,存在多种mapping形式,其中最为稳定的一种是:

如图所示,两个环只map四个原子,稳定性最好。如果将五元环的五个原子都map到六元环上,模拟崩溃的风险会大幅上升。
7. 常见问题
| 小贴士: 如果有这里没有提到的问题,也欢迎随时联系我们提问!您可以直接联系与您对接的销售,也可以把问题发送至邮箱help@dp.tech联系我们。 |
FEP 与 MM-GB/PBSA 原理上的差别是?
FEP 通过分子动力学模拟和自由能微扰方法直接计算两种状态之间的自由能差(ΔΔG),通过热力学路径消除了能量最小化过程中的偏差。MM-GBSA 和 MM-PBSA 则基于单点能计算,将分子置于广义 Born 或 Poisson-Boltzmann 溶剂模型中进行能量估算。相比之下,FEP 更精确,但计算成本更高,而 MM-GB/PBSA 更适合快速筛选。
当蛋白有多条氨基酸链时,应该怎么决定保留哪几条?
通常情况下,只需保留与配体分子有相互作用的氨基酸链即可,删除其他链以提高计算效率。然而,需要注意的是,配体结构的变化可能导致原本不参与相互作用的氨基酸链重新产生相互作用。因此,建议在计算前全面检查所有配体与所有氨基酸可能形成的相互作用,以确保最终保留的氨基酸链能够准确反映系统中的关键相互作用。
在做蛋白准备的时候,要保留蛋白晶体结构中的水吗?
通常建议保留以下几类水分子:(1) 能够与蛋白-配体形成关键相互作用的水分子,例如参与氢键或其他非共价相互作用的水分子;(2) 对蛋白局部区域稳定性有显著贡献的水分子,例如维持活性位点或关键次级结构的水分子。对于其他不重要的水分子,可以删除以简化系统并提高计算效率。在保留水分子前,建议结合晶体结构和模拟需求进行综合判断。
使用 FEP 计算膜蛋白体系时,有哪些注意事项?
对膜蛋白体系进行 FEP 计算时,为了尽可能保留体系的生物环境,计算需在 POPC 构建的膜环境下进行。因此,在蛋白处理时需要对蛋白加膜,否则可能会影响计算结果的准确度,甚至导致任务失败。此外,由于带膜体系的原子数较多,再加上大体系动力学模拟的平衡需求,整体计算量通常是水溶性蛋白的 2~3 倍。因此,任务运行时间会相应延长,需合理规划计算资源。
FEP 计算时,对带电小分子需要做什么特殊处理吗?
如果参与 FEP 计算的配体净电荷相同(如均为 0),则按照普通的 FEP 流程处理,只需确保每个配体的化学结构和电荷准确即可。如果配体的净电荷发生变化(如净电荷变化为 1),Uni-FEP 会调用特殊算法,确保体系总电荷在分子动力学模拟过程中保持平衡。这通常通过将体系中的水分子逐渐变为正、负离子来实现。同时,适量添加阴、阳离子的盐溶液可以进一步改善计算的稳定性。
分子活性数值与结合自由能的换算关系是什么样的?
分子活性(如 IC₅₀、Kᵢ 或 Kd 值)与结合自由能(ΔG)的关系可通过以下公式(1)近似计算。其中 R 为气体常数(0.001986 kcal/(mol·K)),T 为温度(通常为 298 K)。通过此公式,可以将活性指标转换为对应的 ΔG 值。需要注意的是,ΔG 的每 1 kcal/mol 的变化,通常对应活性变化约 6 倍。因此,结合自由能的微小差异可能对活性产生显著影响。在使用此公式时,应确保所使用的活性指标(如 IC₅₀ 或 Kd)在相同的实验条件下测得,且为非细胞水平的实验值,否则换算可能会存在误差。通过这种转换,ΔG 值可以与计算结果直接对比,从而更好地评估模型的预测准确性。
一般来说,RMSE 值和 R² 结果在什么范围内可以说明 FEP 的计算结果比较好?
当分子的活性实验值跨度较大时,建议参考 R² 值。R² 越接近 1,说明计算结果越准确,通常 R² 大于 0.4 的结果可以接受。当分子活性值较接近时,建议参考 RMSE �值。RMSE 越小,说明计算结果越好,一般来说,FEP 的 RMSE 值小于 1.4 kcal/mol 即为可接受范围。
如何对结果中的热力学循环进行解读?
热力学循环是通过 ΔΔG 的计算形成闭环。例如,若提交了 3 个分子,形成了闭环 A→B、B→C、C→A,根据热力学原理,理论上热力学循环误差应为 0。因此,热力学循环误差的绝对值越小,说明该循环内涉及的配对计算结果的可靠性越高。较大的热力学循环误差可能表明体系存在计算偏差或不平衡,需要进一步检查体系构建或模拟参数。
FEP是否可以用于预测激动活性?
结合自由能是激动活性的必要不充分条件。激动剂首先需要稳定地结合到靶蛋白上(表现为较低的结合自由能,ΔG),然后小分子结合后诱导特定的构象变化,从而触发信号传导。总而言之,结合是激动活性的前提条件,因此,高活性的分子至少应当表现出良好的结合能力。
Uni-FEP的分子相似性是如何定义的?
基于合理的映射关系,分子相似性通过以下维度进行综合评估:分子间的电荷分布、核心区域的构象差异、环系的空间匹配、成键/断键的变化,以及虚拟原子区域的特征。分子相似性直接反映微扰对的计算成功率。其中,similarity score < 0.001 的分子对不能被连接和计算。
Uni-FEP的副本数设置为Auto的时候,是依据什么调整的?
自动副本数设置主要依据变化类型决定。其中,电荷变化、骨架跃迁、较大R-group的消失都会增加lambda数量,会在16, 20, 24三个值里根据规则调整。
上传分子后,更新微扰分子对信息(Update Pairs)时等待时间过长,该如何处理?
在更新微扰分子对信息时,Uni-FEP 会计算任意两个分子之间的 3D 原子映射(Atom Mapping)并评估其结构相似性。因此,计算时长通常随上传的分子数量呈现平方关系增长。此外,若分子间结构差异较大或叠合程度较差,也会显著延长该计算过程。当用户发现等待时间异常时,建议检查分子间的结构叠合情况,确保分子结构已合理对齐后再重新更新微扰分子对信息。
FEP模拟过程的任务失败一般是什么原因?
Hermite® Uni-FEP 在任务执行前嵌入了大量前置检查,能有效规避大部分错误情况。因此,FEP 模拟任务失败绝大多数归因于 3D 原子映射(Atom Mapping)不合理。常见问题之一是,当两个分子在骨架结构或官能团类型上存在显著差异时,原子映射关系往往变得过于复杂,进而引发模拟过程中的采样不足或计算崩溃。
为解决此类问题,建议在 Uni-FEP 中采用手动或自动生成“过渡分子”的策略,将大幅度结构变化拆分为多个较小改动。例如,当 A 分子到 B 分子在两个官能团(如 R1 和 R2)上均发生变化时,直接转换可能导致体系瞬间剧烈波动,不利于采样。此时,可以先构建中间分子 C,仅将 A 分子的 R1 更换为 B 分子的 R1(同时保持 R2 不变);随后,在分子 C 的基础上,再将 R2 更换为 B 分子的 R2。这样分步进行 A→B 的转换,能够降低映射复杂性,改善采样效果,从而显著提高计算准确性并降低风险。
在 Uni-FEP 中,用户可自行创建并上传这些中间分子,使软件分段完成自由能计算,确保整体模拟过程更加稳定、收敛性更好。
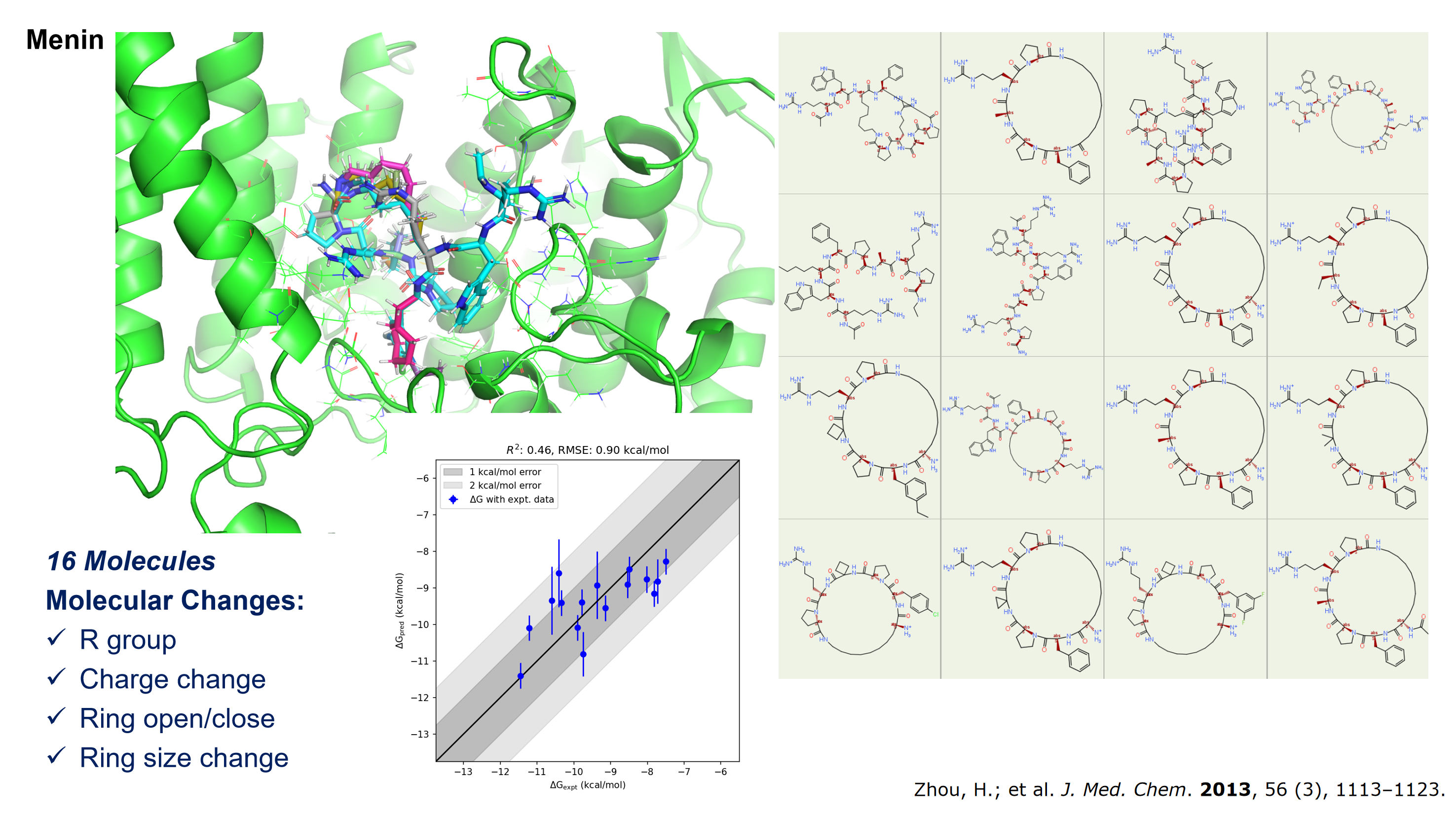
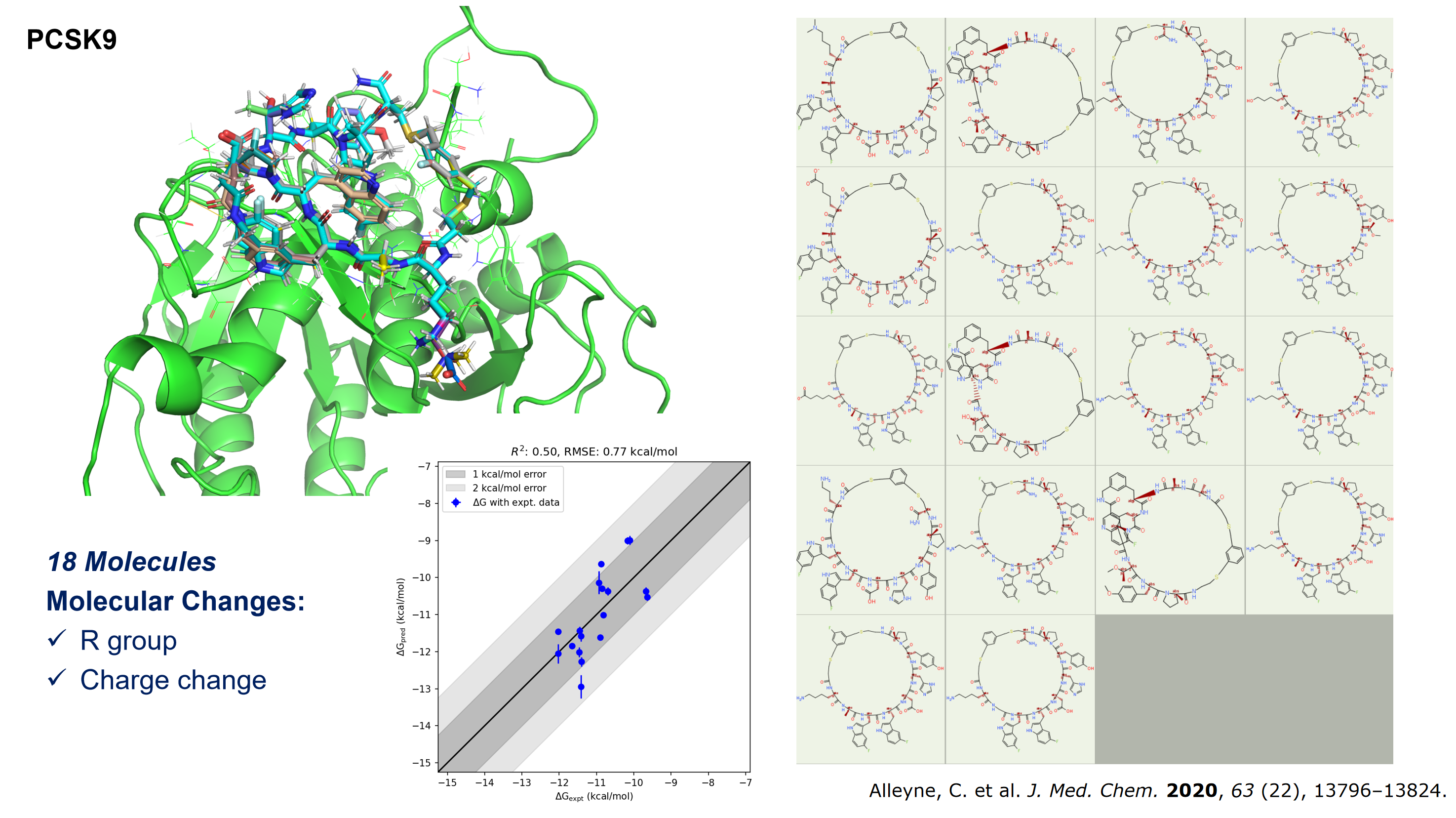
针对多肽、环肽等肽类似物进行 FEP 计算时,是否有专门的分子力场?
在药物设计中,肽类似物由于可能包含非标准氨�基酸等特殊结构,常常被视为小分子处理。为此,我们推荐使用适用于小分子的分子力场进行计算。近期,我们已在 Menin(包含16个分子)和 PCSK9(包含18个分子)两种环肽体系上对 Hermite® Uni-FEP 进行了测试,结果显示能有效应对包括大侧链基团变化、电荷变化、环大小变化以及环断裂等较为复杂的情形。
8. 基准测试
基准测试旨在系统性地评估自由能微扰(FEP)方法在药物研发中的性能,验证其在不同场景下预测结合自由能的准确性、稳定性和适用性。与实际项目中的验证阶段类似,基准测试使用经过广泛验证和公开报道的标准数据集,通过统一的测试流程和评价指标,展示方法在不同靶点和化学变换场景中的整体表现。
在结果解读方面,当分子活性的实验值跨度较大时,R²(决定系数) 更适合作为评价指标,R² 越接近 1,说明预测结果与实验数据的相关性越强,通常 R² > 0.4 被认为是可接受的。当分子活性值较为接近时,RMSE(均方根误差) 更具参考价值,较低的 RMSE 代表更小的预测误差,通常 RMSE < 1.4 kcal/mol 可以视为较好的预测性能。
通过这些基准测试,我们能够全面了解 FEP 方法在不同体系和场景下的表现,识别可能的局限性,并为后续的算法优化和药物研发提供科学依据。
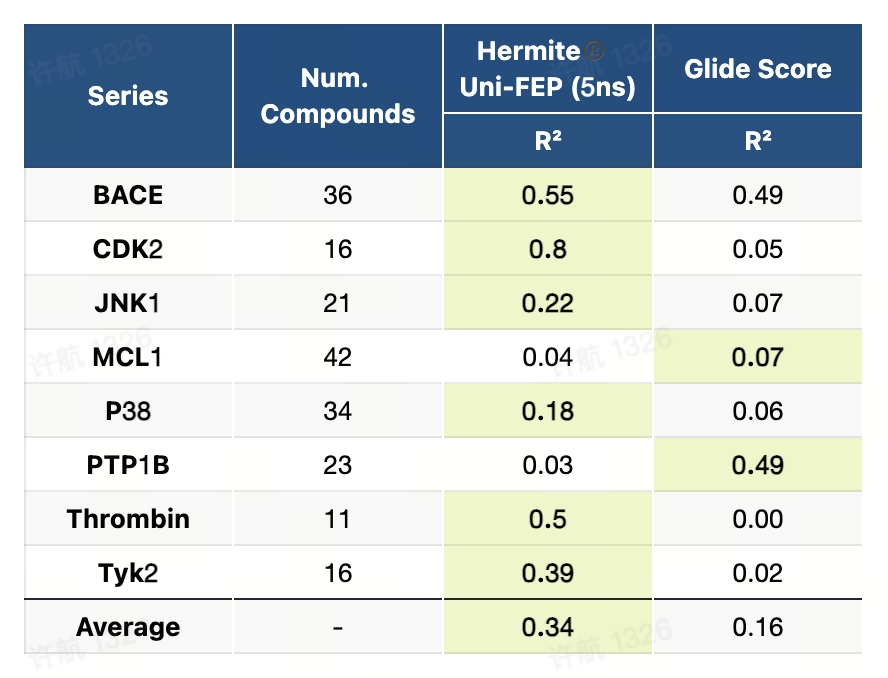
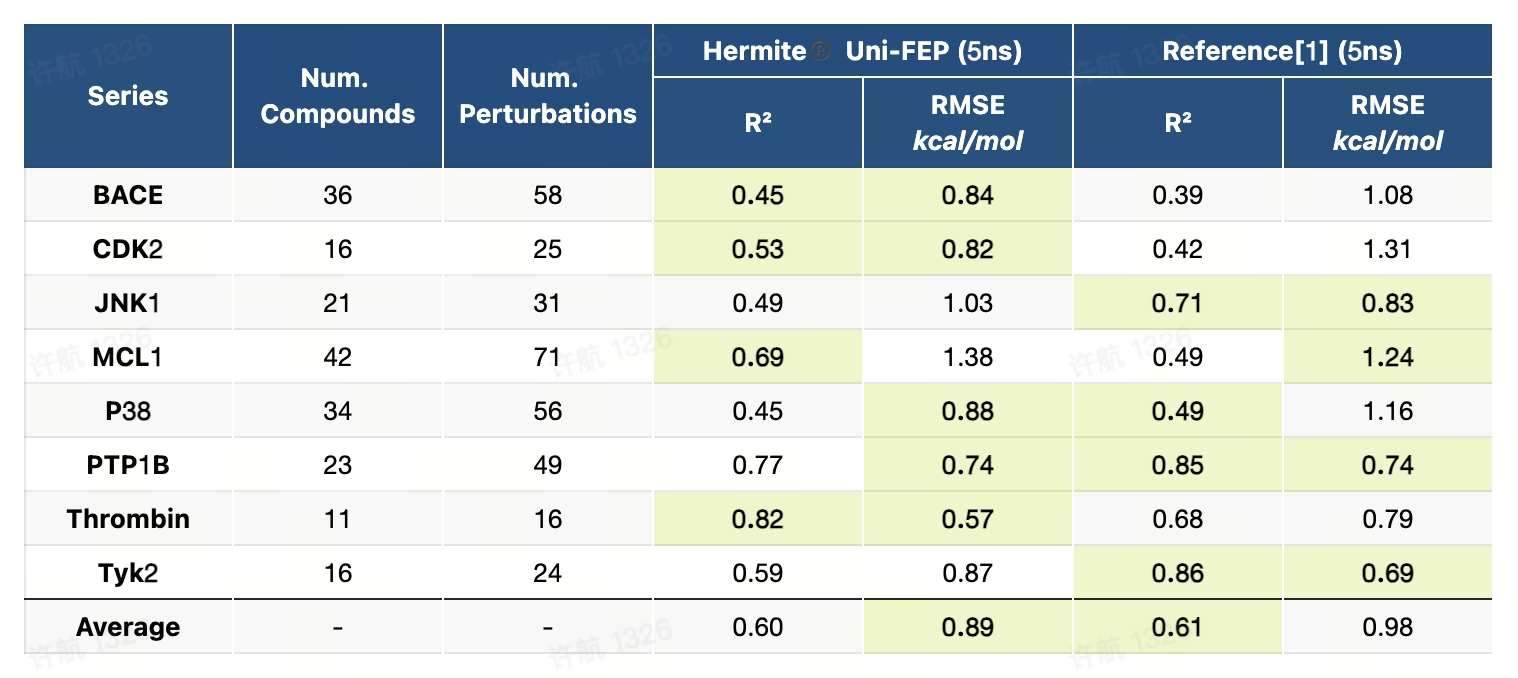
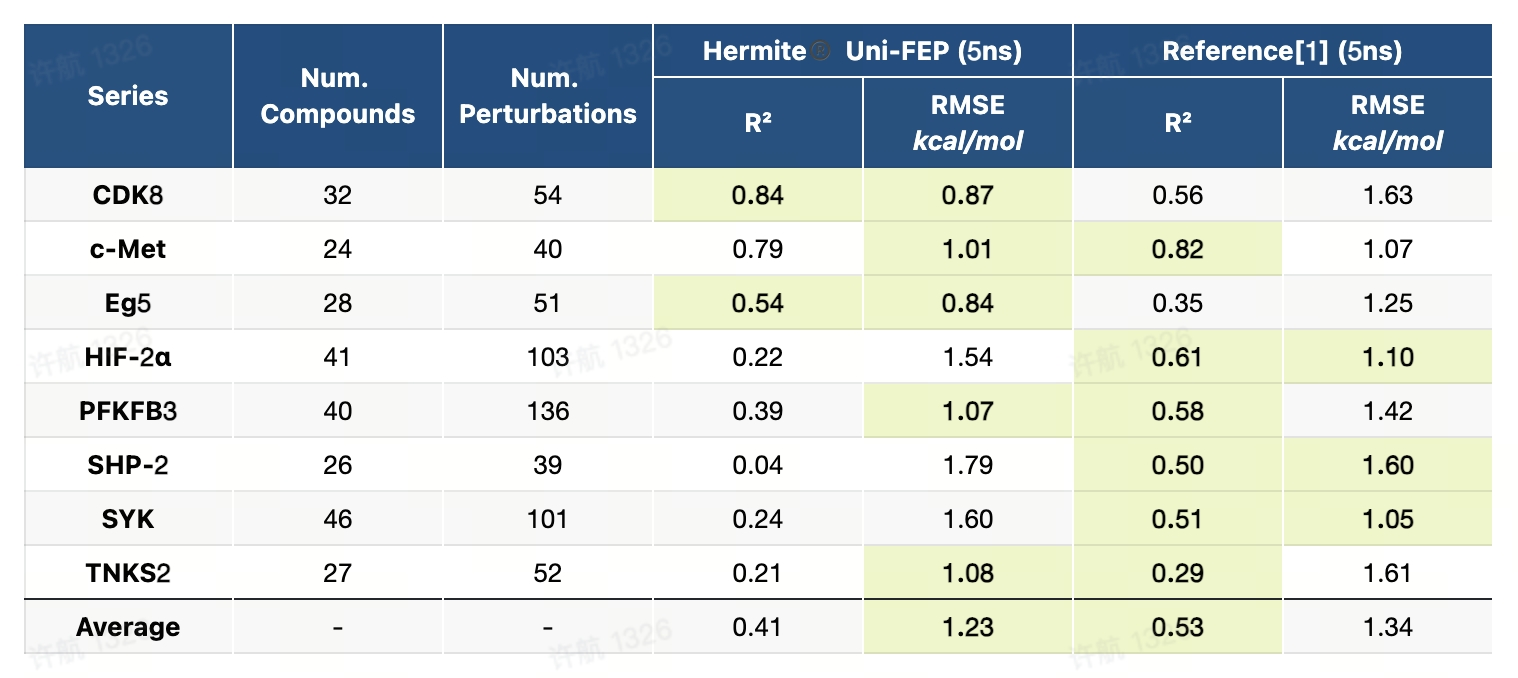
8.1 JACS-2015基准测试(RBFE)
JACS-2015基准测试是一个高精度且系统化的数据集,专为评估自由能微扰(FEP)方法在药物研发中的广泛适用性而设计。该基准测试覆盖了8个药物开发中的经典靶点,包括蛋白激酶、酶类和受体,设计了8类化学变换场景,如电荷变化、骨架跃迁、环结构转换等,涵盖了药物设计中具有挑战性的典型问题。测试流程提供了详尽的蛋白-配体准备指南、配体建模方法以及标准化的计算设置,为研究者在真实场景下评估和优化FEP方法提供了全面的支持。
8.2 JCIM-2020基准测试(RBFE)
JCIM-2020基准测试是一个设计精良且全面的数据集,专注于评估自由能微扰(FEP)方法在复杂药物设计场景中的性能。该基准测试选择了8个具有代表性的药物靶点,并包含264个配体,设计了包括电荷变化、环结构转换、骨架跃迁等化学变换场景,以模拟多样化的药物研发需求。测试提供详细的蛋白质结构准备和配体建模流程,旨在通过系统验证,推动FEP方法在药物开发中的实际应用。
8.3 GPCR-FEP基准测试(RBFE)
GPCR-FEP基准测试是目前针对G蛋白偶联受体(GPCR)的最全面的数据集之一,专为评估自由能微扰(FEP)方法在膜蛋白靶点中的表现而设计。该基准测试覆盖了8种GPCR靶点,构建了12个数据集,共139个配体,并提供了详细的蛋白-配体结构准备、水分布方案和膜环境构建方法。通过精准再现膜蛋白体系的复杂性,该基准测试为研究者提供了高质量的数据和标准化的计算框架,推动FEP技术在GPCR靶点药物研发中的应用。
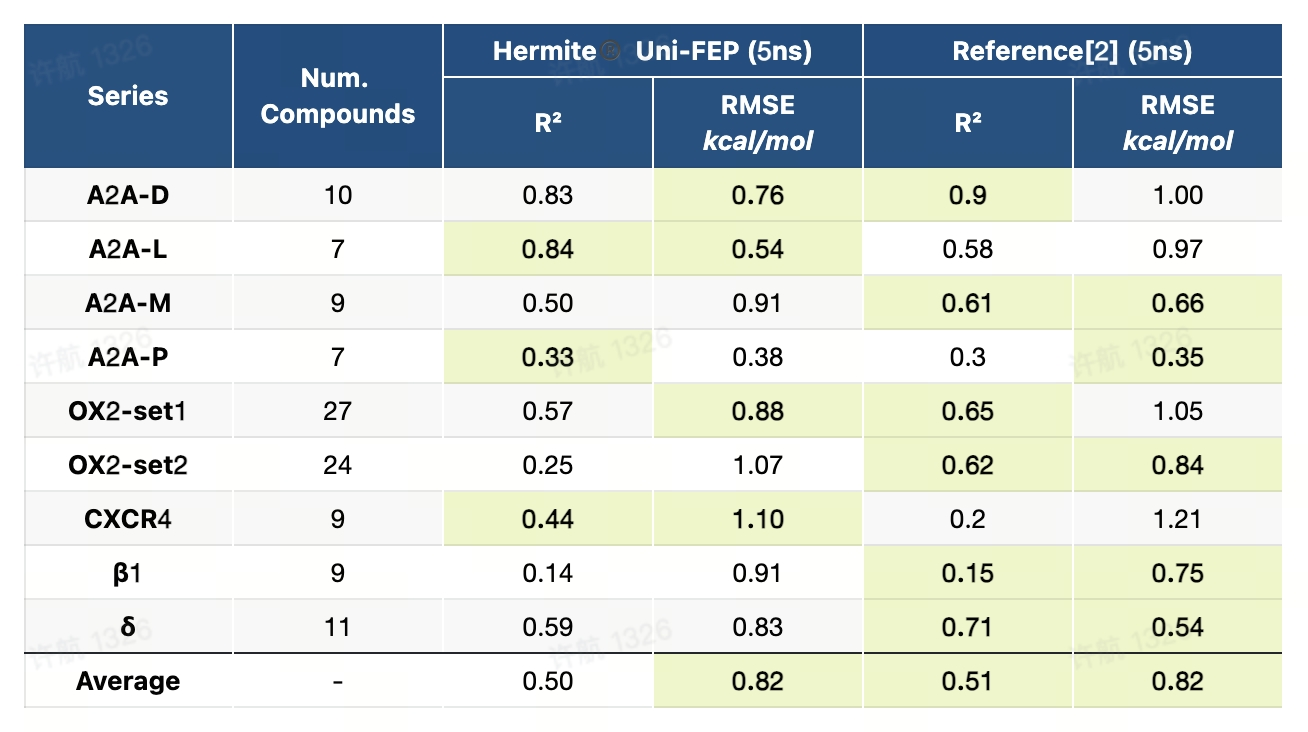
| Reference: [1] Ross GA, Lu C, Scarabelli G, Albanese SK, Houang E, Abel R, Harder ED, Wang L. The maximal and current accuracy of rigorous protein-ligand binding free energy calculations. Communications Chemistry. 2023 Oct 14;6(1):222. [2] Guo Y, Zhou Y, Bai Q, Bo Z, Song K, Chang J, Zhang Y, Yang M, Deng Y, Wang D. A Benchmark for Accurate GPCR Ligand Binding Affinity Prediction with Free Energy Perturbation. ChemRxiv, 2023. |
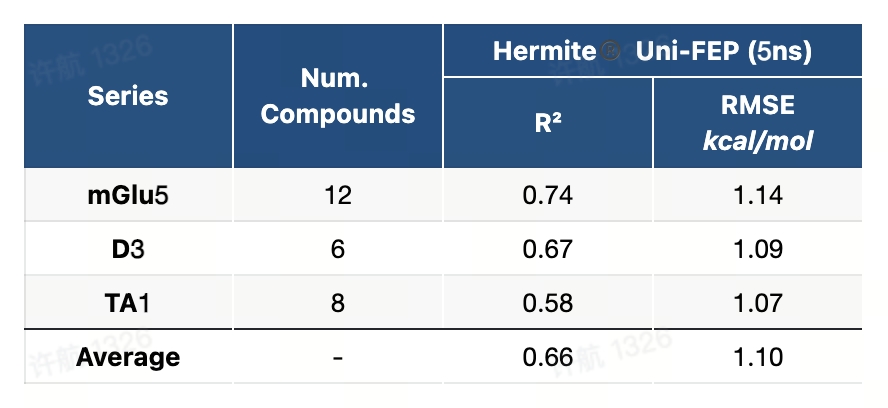
8.4 JACS-2015基准测试(ABFE)
JACS-2015基准测试是一个高精度且系统化的数据集,专为评估自由能微扰(FEP)方法在药物研发中的广泛适用性而设计。该基准测试覆盖了8个药�物开发中的经典靶点,包括蛋白激酶、酶类和受体,设计了8类化学变换场景,如电荷变化、骨架跃迁、环结构转换等,涵盖了药物设计中具有挑战性的典型问题。测试流程提供了详尽的蛋白-配体准备指南、配体建模方法以及标准化的计算设置,为研究者在真实场景下评估和优化FEP方法提供了全面的支持。
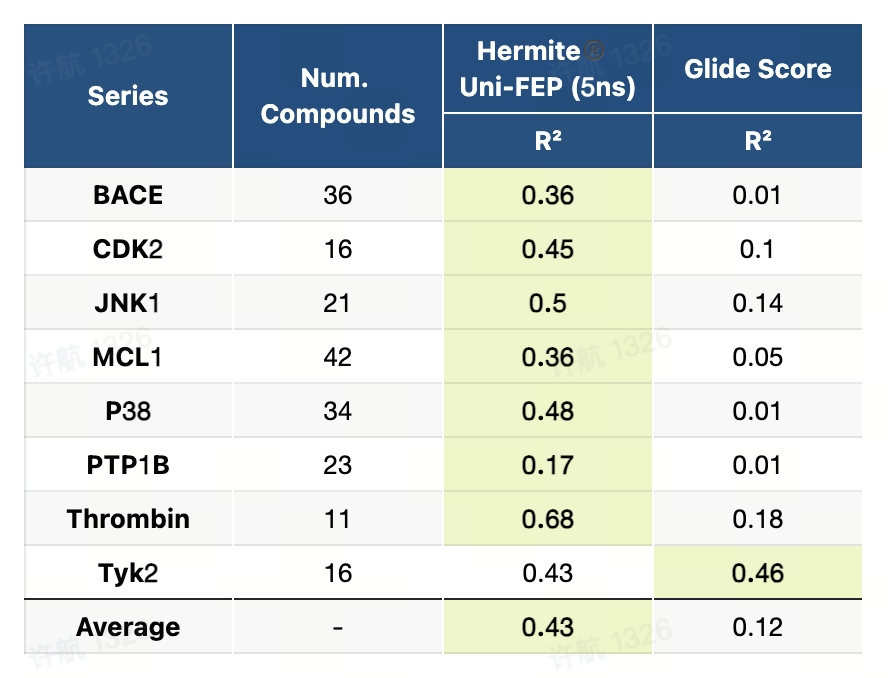
8.5 JCIM-2020基准测试(ABFE)
JCIM-2020基准测试是一个设计精良且全面的数据集,专注于评估自由能微扰(FEP)方法在复杂药物设计场景中的性能。该基准测试选择了8个具有代表性的药物靶点,并包含264个配体,设计了包括电荷变化、环结构转换、骨架跃迁等化学变换场景,以模拟多样化的药物研发需求。测试提供详细的蛋白质结构准备和配体建模流程,旨在通过系统验证,推动FEP方法在药物开发中的实际应用。