Induced Fit Dcoking
1. Introduction
The lock-key model was initially proposed for the binding conformation of protein-ligand, that is, the conformation of protein and ligand does not change before and after binding, just like a key and a lock; However, in actual biochemical processes, the intermolecular forces generated when ligands bind to pockets will induce conformational changes, which is called Induced Fit effect. At the same time, the influence of water on protein and ligand binding should also be considered. Uni-IFD accurately predicts the binding mode of drugs and targets by simulating the “induced fit” effect when drug molecules bind to targets.
2. How to use
2.1 Entrance
- Left general menu bar Menu → Function → Virtual Screening → Induced Fit Docking.
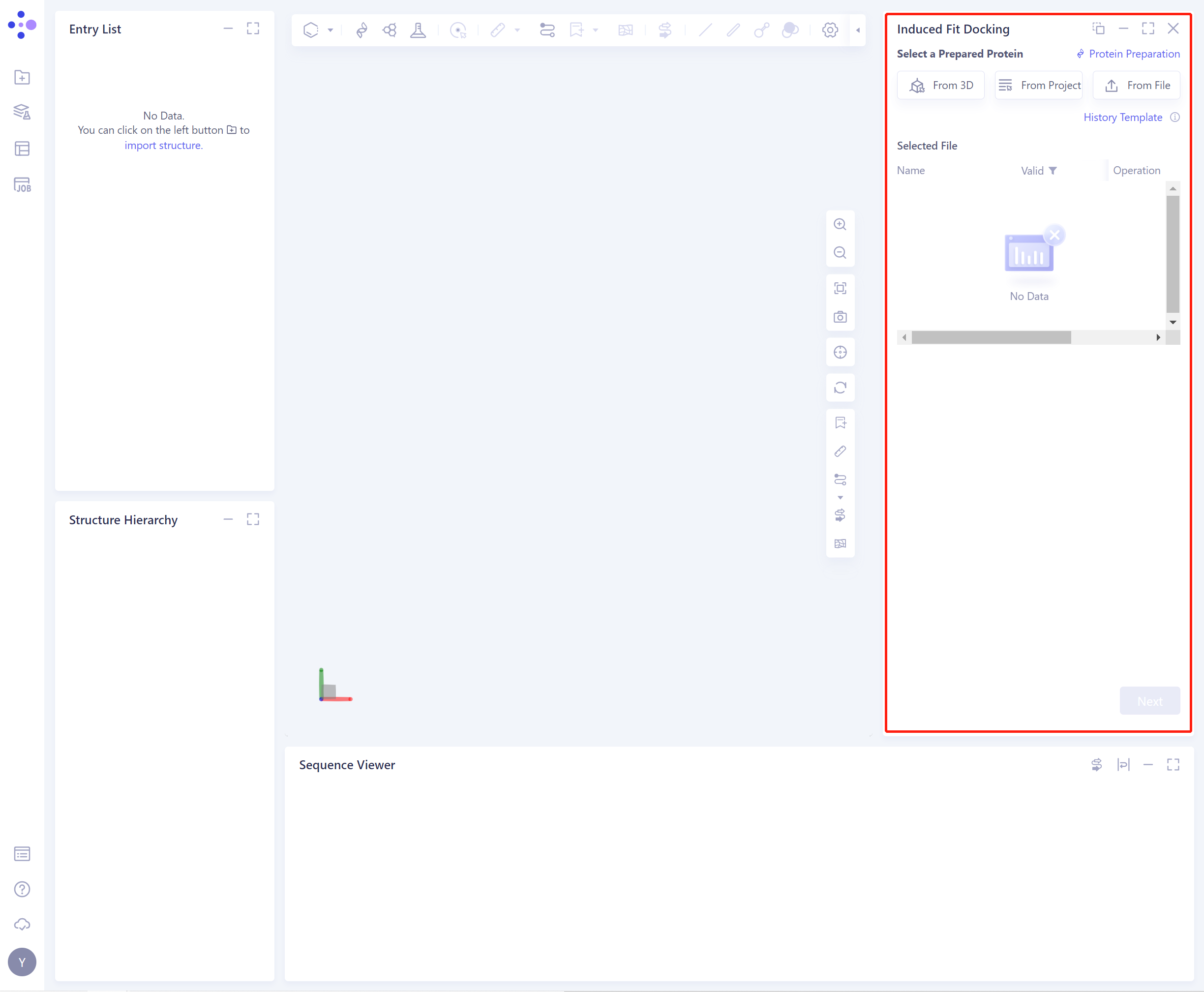
- The operation box of the Induced Fit Docking (shown in the red box) appears on the right side, and the overall interface is as follows:
2.2 Operation
-
There are three ways to introduce protein structure:
-
1)From 3D Works pace: Select proteins from graphical components. 3D Workspace only supports the selection of protein level, which can be selected in Hierarchy (linkage 3D).
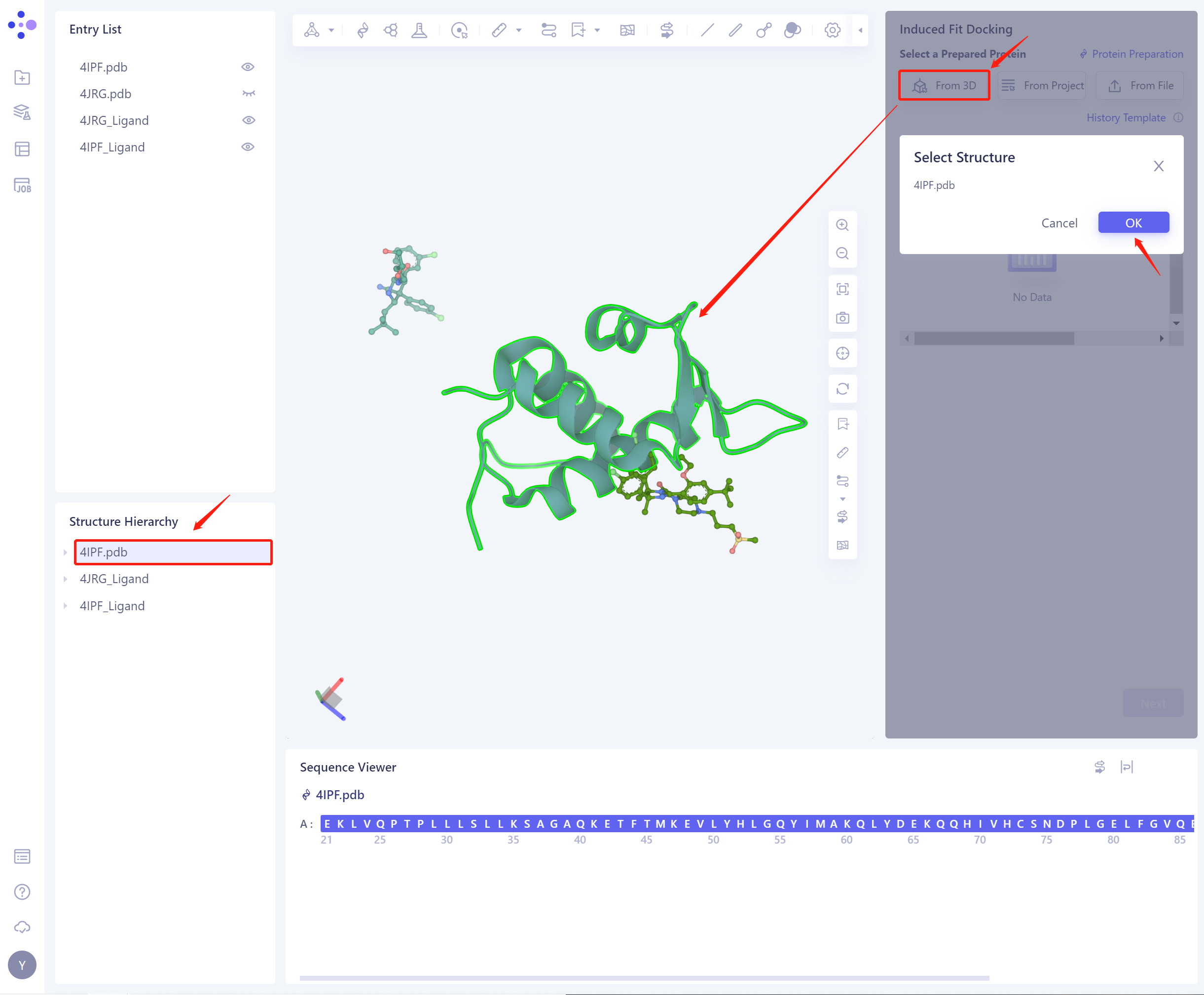
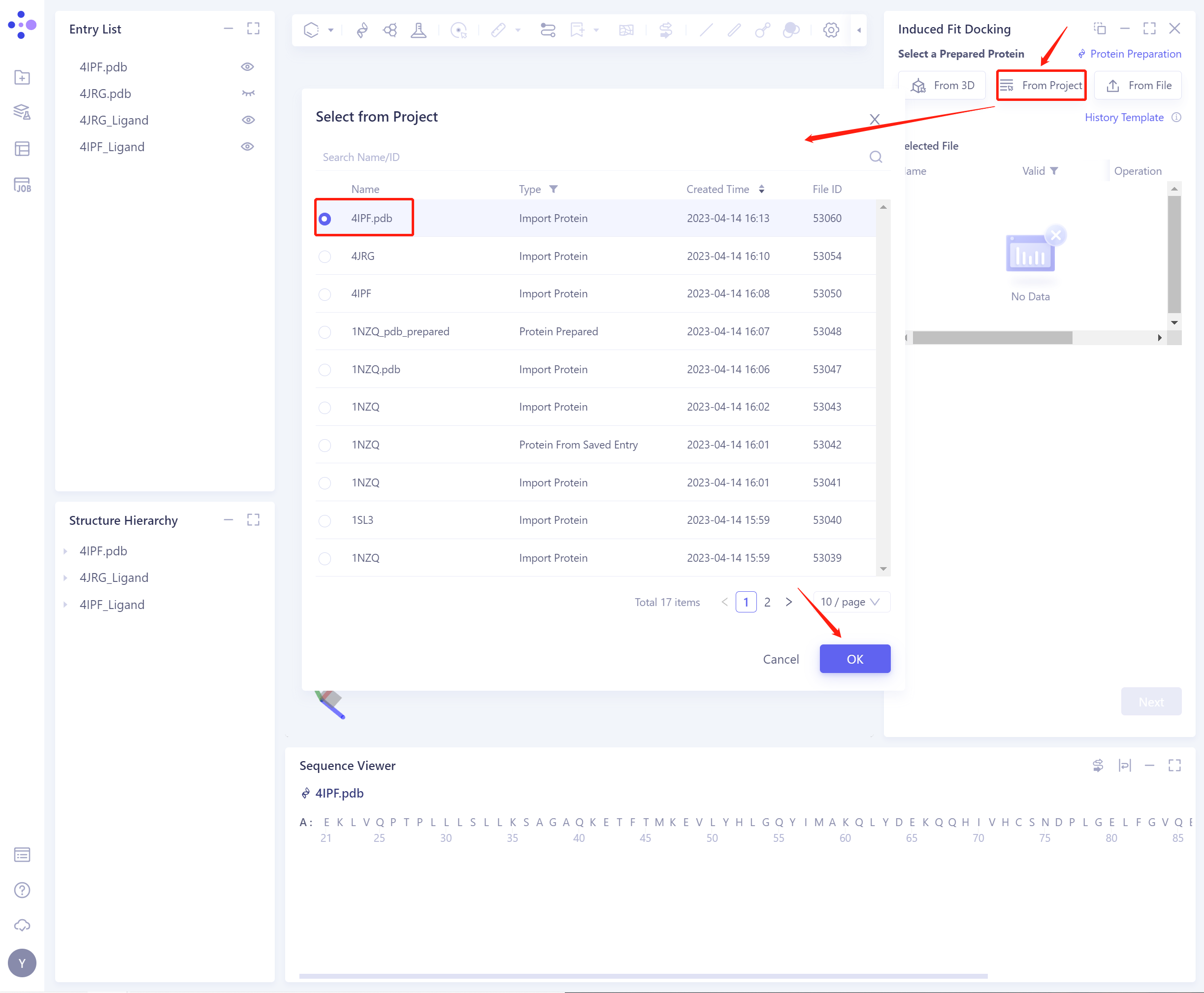
- 2)From Project: Select proteins from historical projects.
- In the pop-up Select from Project operation interface, you can search for files by file name and ID through Search Name/ID, and adjust the Type to select files.
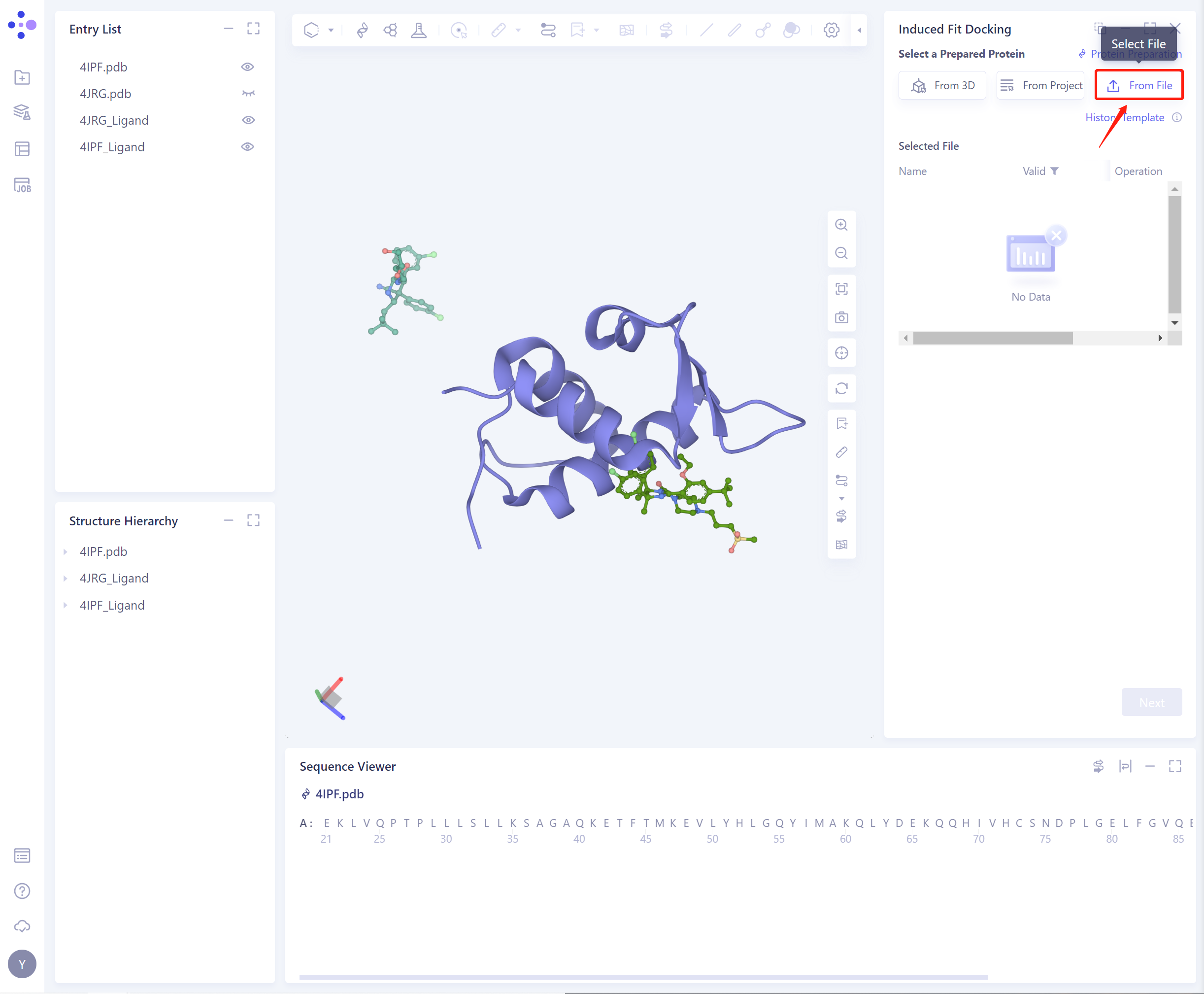
- 3)From File: Upload a local protein file in.pdb format.
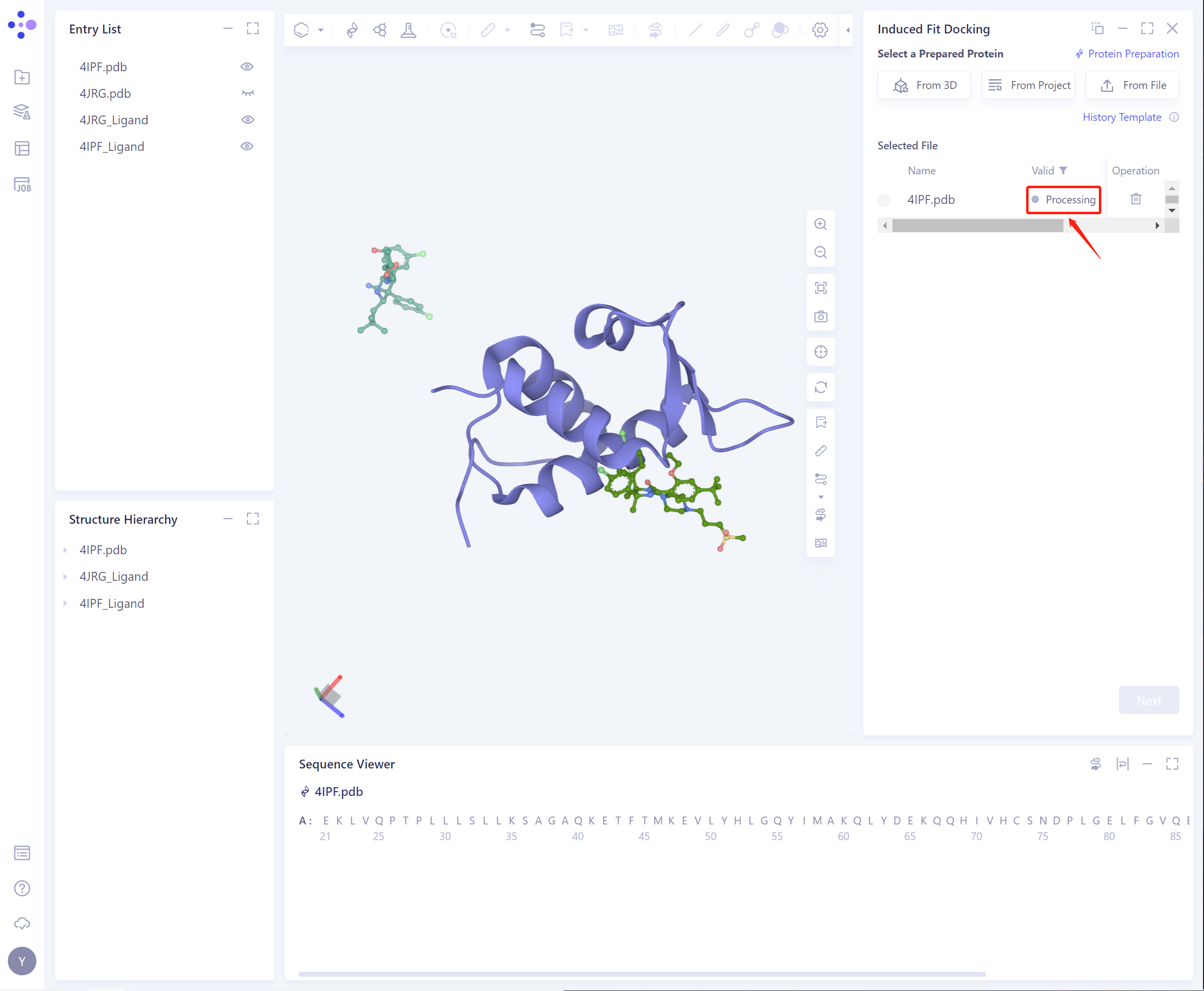
- The selected protein will be checked by the force field, and the following figure shows the checking.
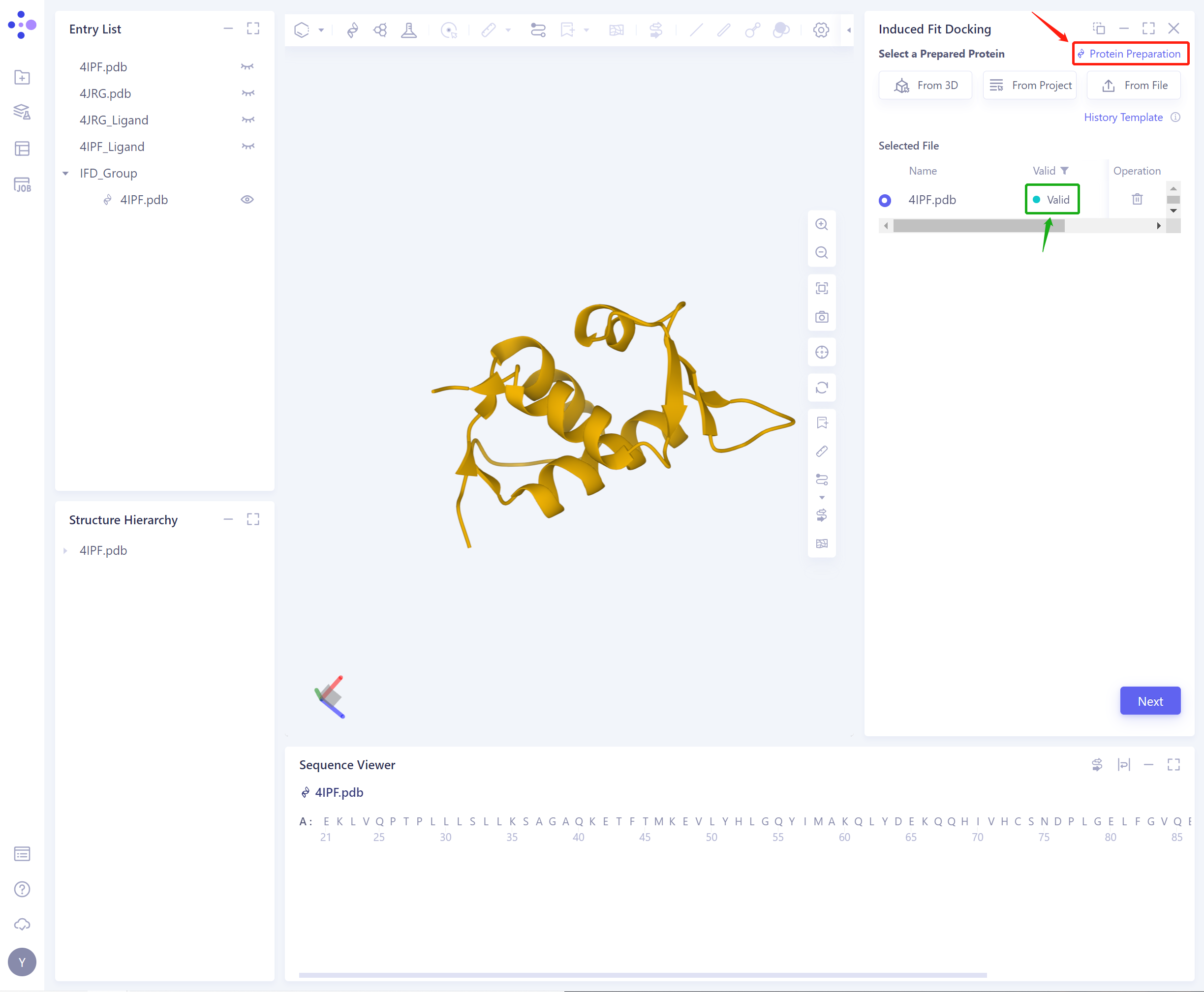
- If the protein does not pass the force field check (here is Valid, that is, the force field check passes), protein preparation is required (see the red box on the upper right of the figure for the shortcut).
-
After the protein force field check is passed, you can continue to use the function.
-
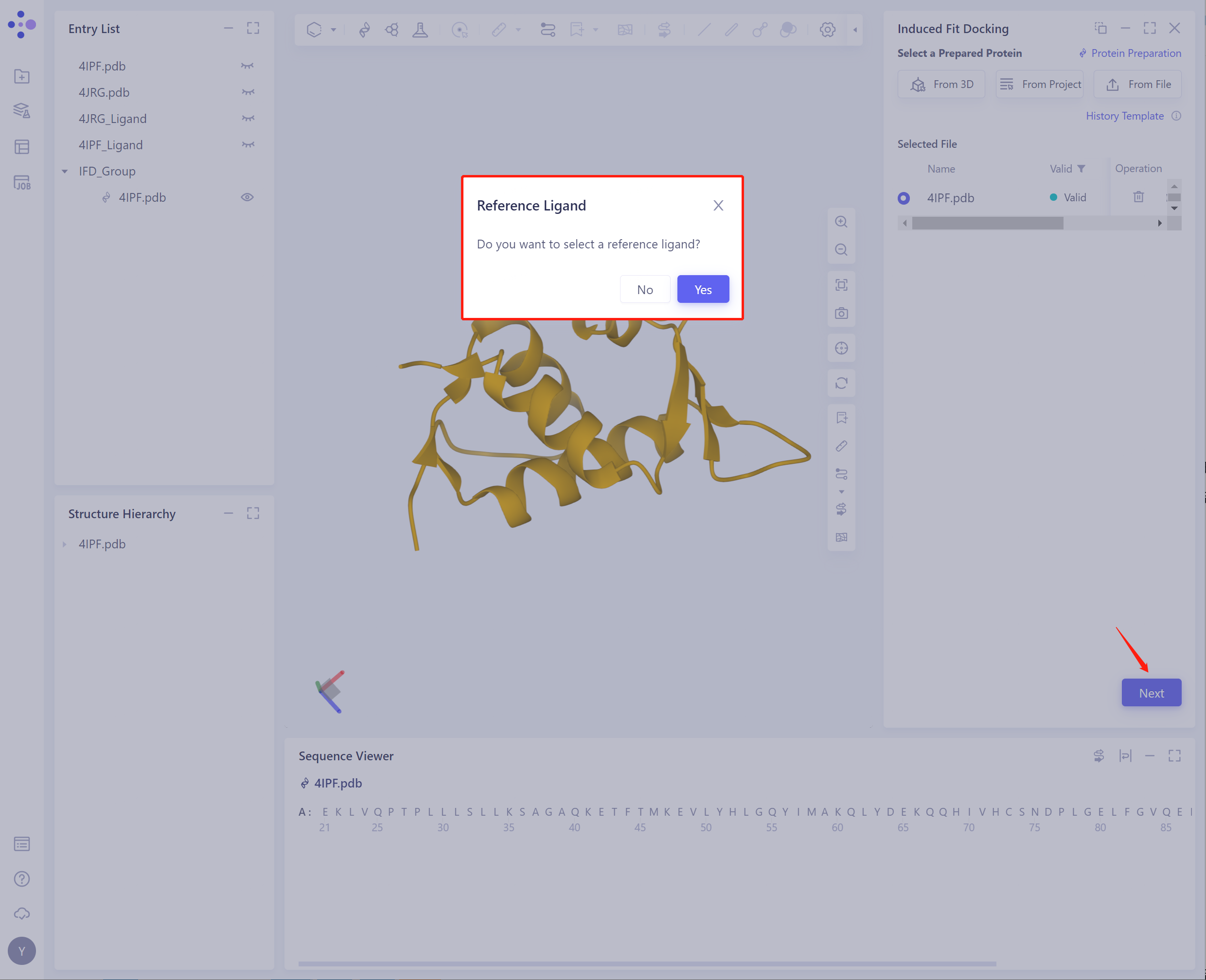
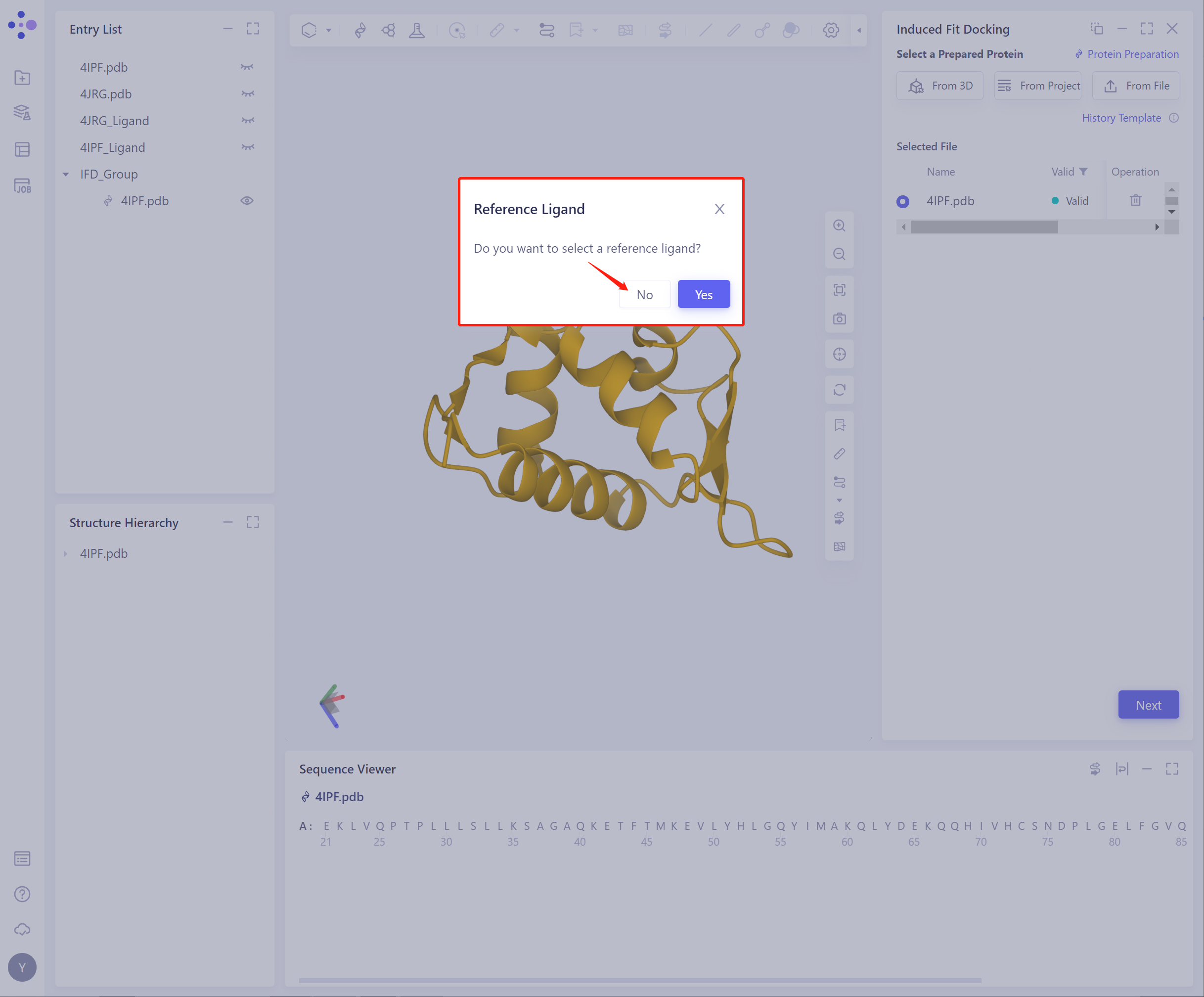
Click Next to prompt whether to select reference ligand:
-
Path 1: Whether to select Reference Ligand: Click Yes to Select a Reference Ligand.
-
There are three ways to import ligand:
-
1)From 3D Works pace: Select the ligand from the graphical component. 3D Workspace only supports the selection of Residue level, which can be selected in Hierarchy (linkage 3D).
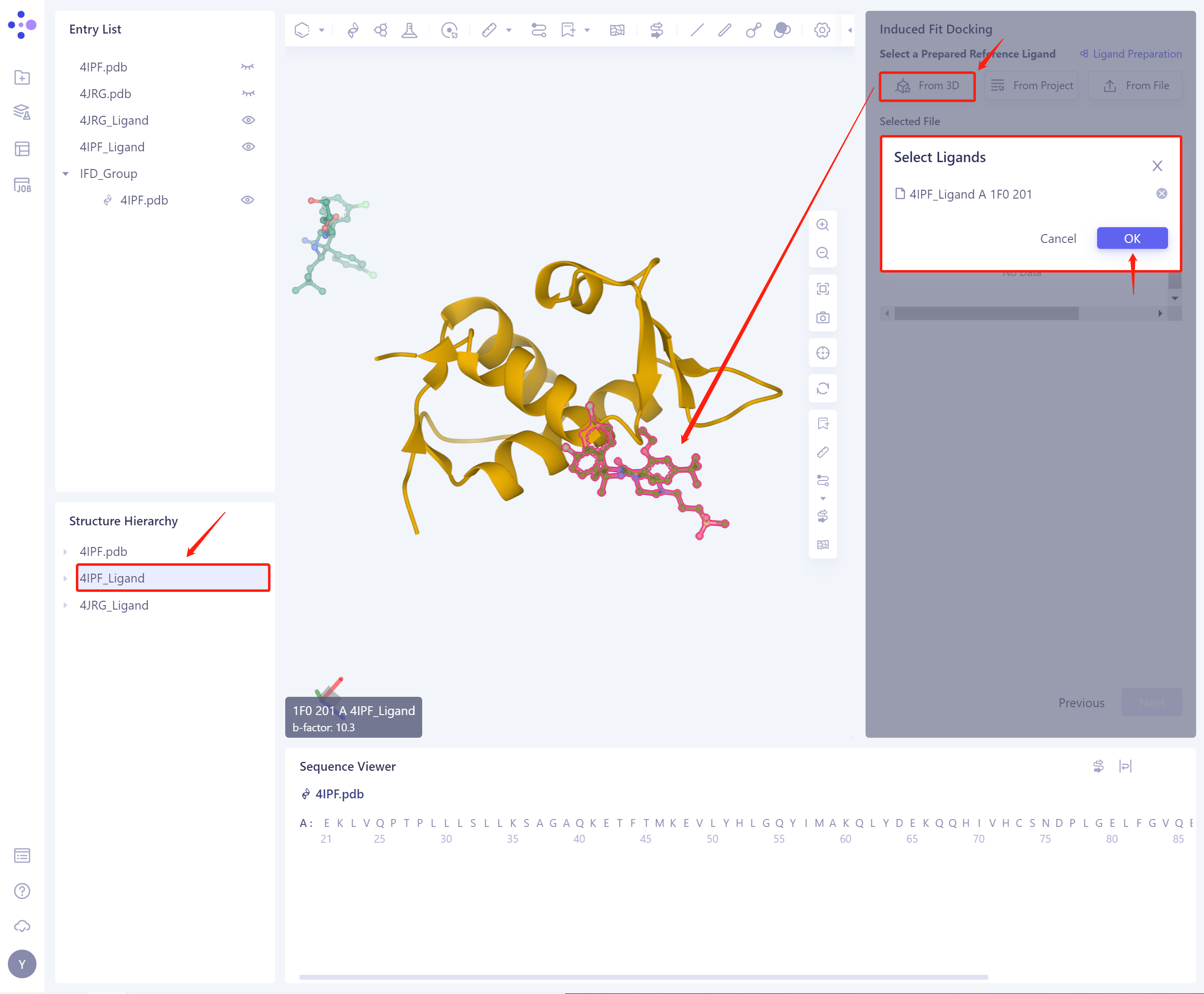
-
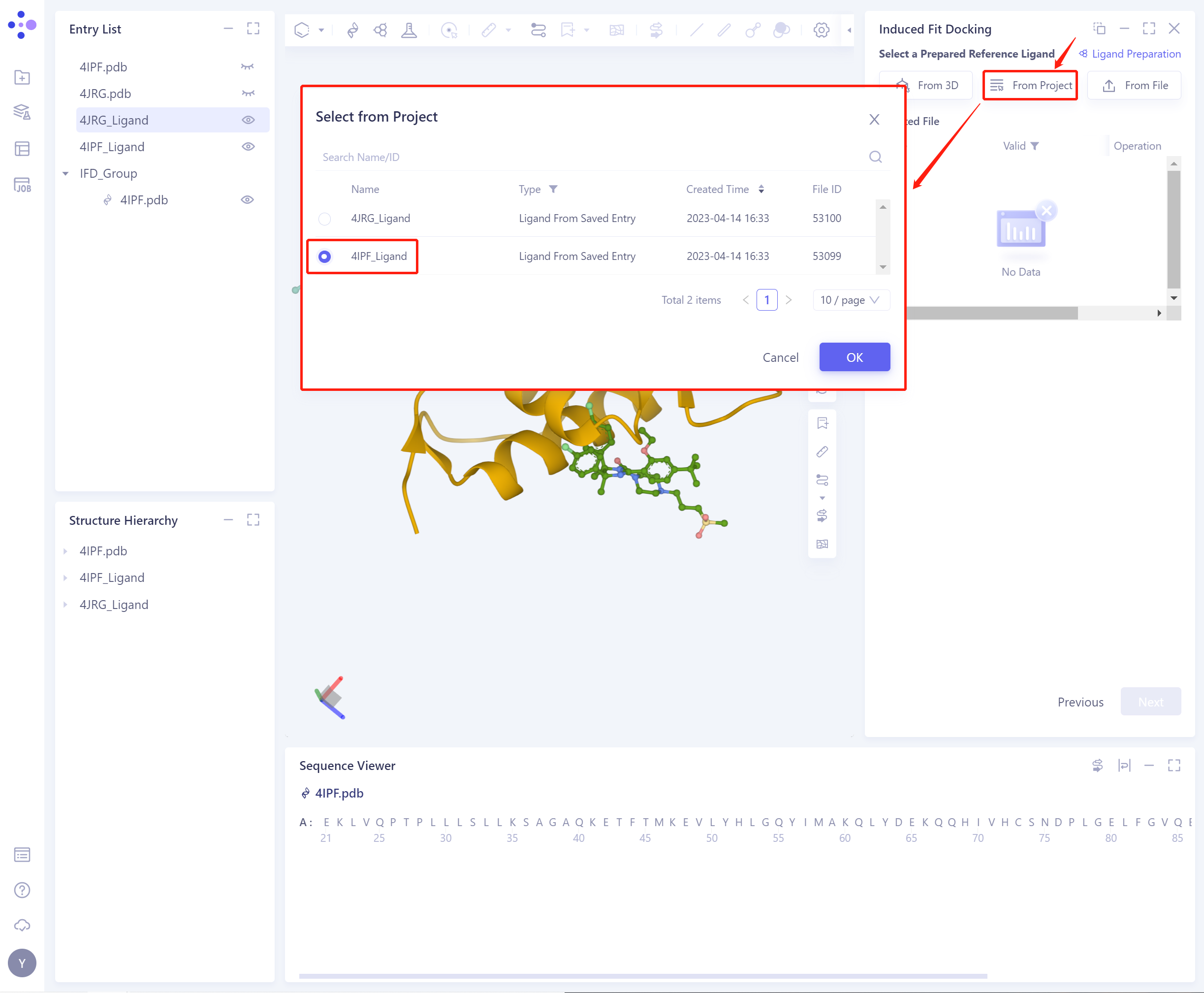
2)From Project: select the ligand from the historical project.
-
In the pop-up Select from Project operation interface, you can search for files by file name and ID through Search Name/ID, and select the ligand file by adjusting the type (Type).
- 3)From File: Upload the local ligand file, which supports the.sdf/.mol format.
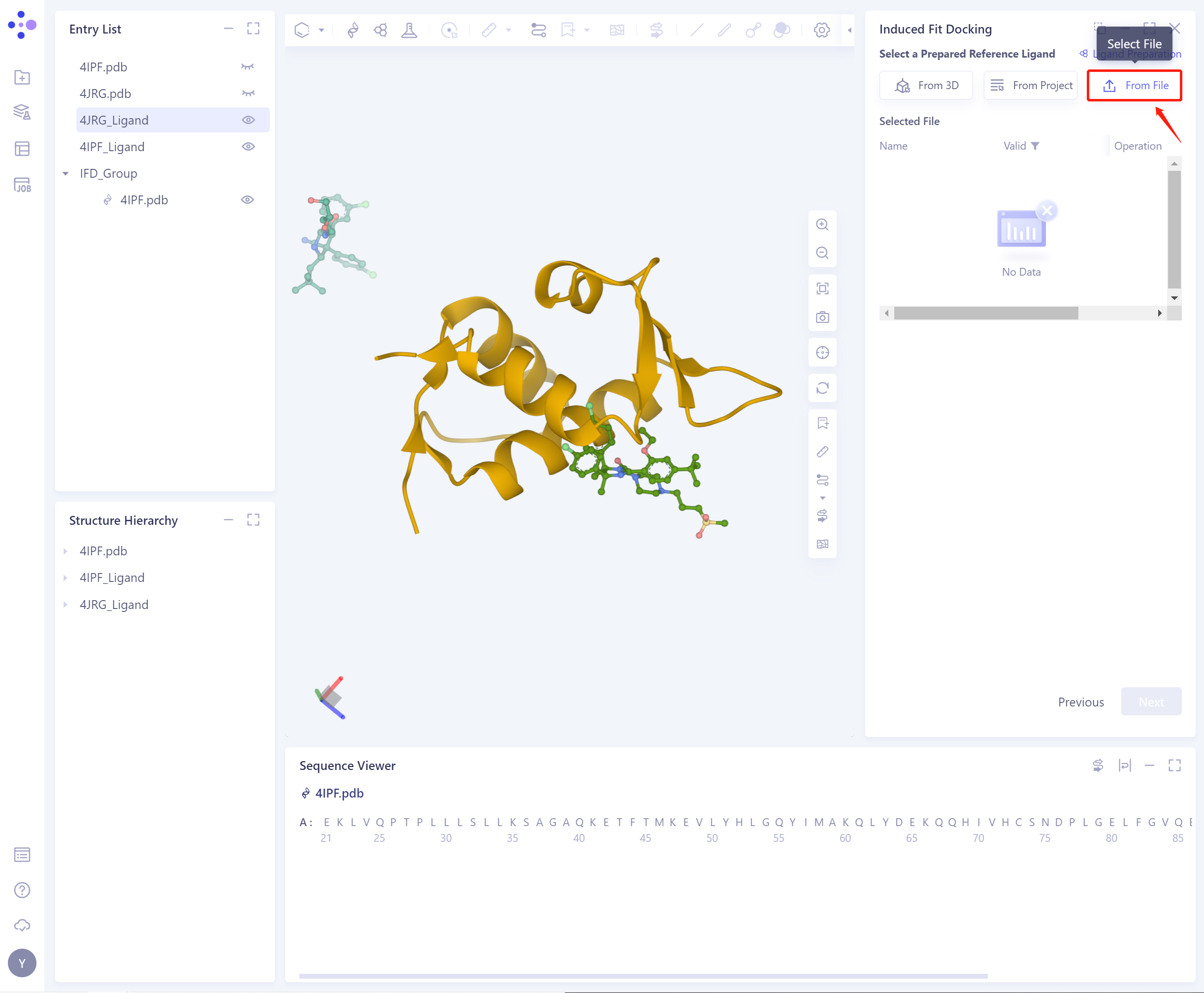
- Before importing the ligand, you need to select whether to repair the bond information that may be lost by the ligand. Yes is selected by default.
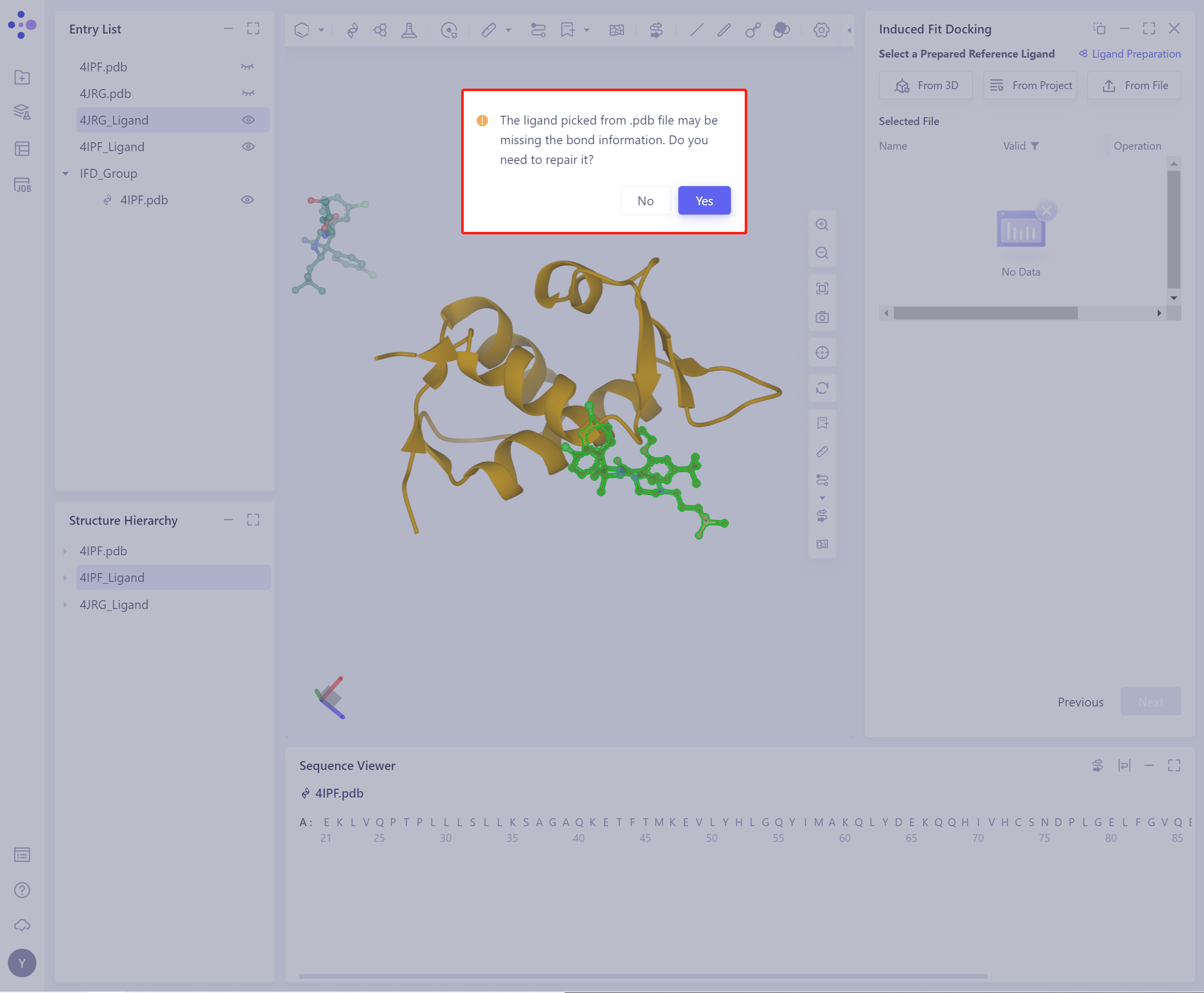
- The selected ligand will be checked by the force field, and the following figure shows the checking.
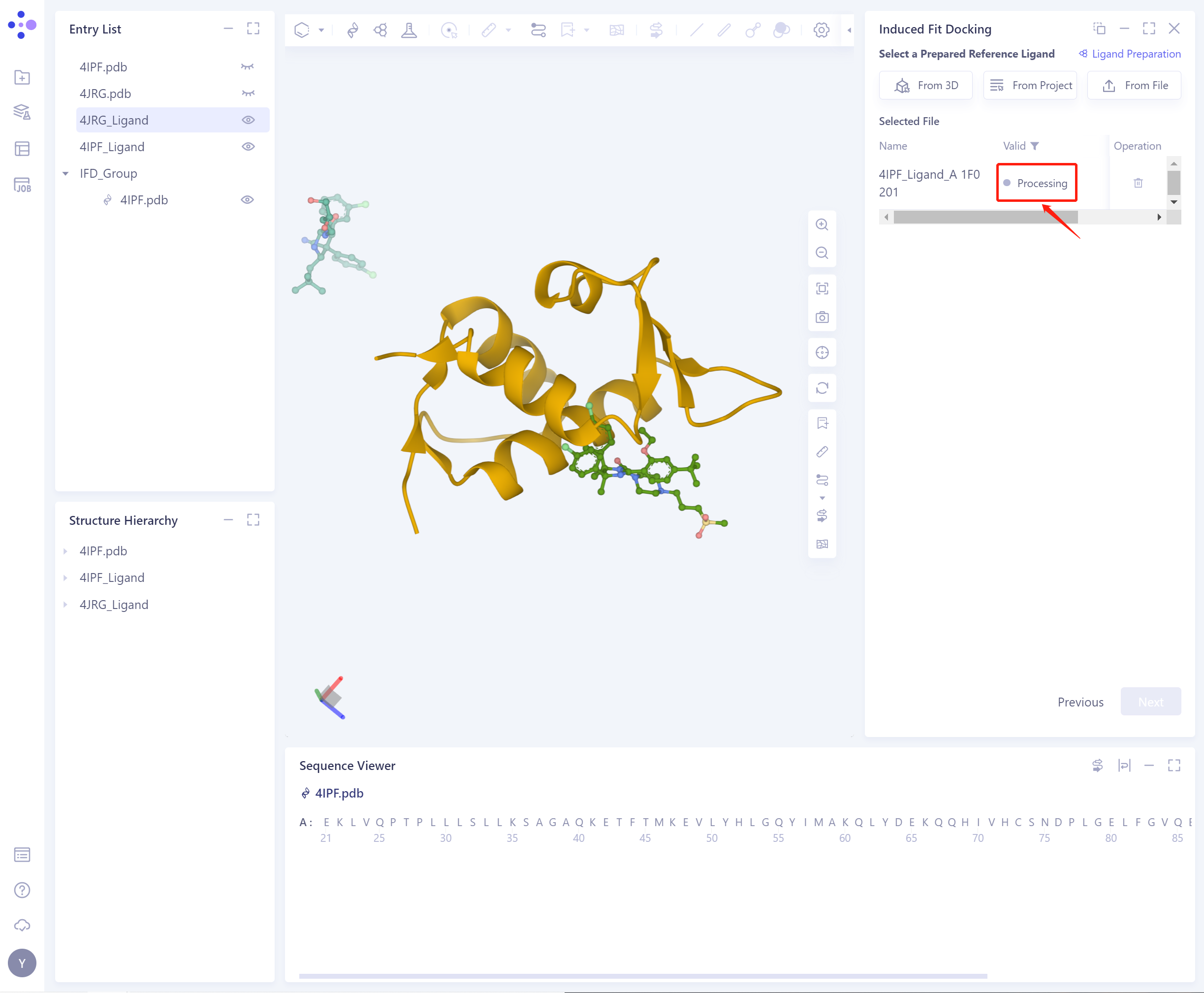
- If the ligand force field inspection is not passed (here is Valid, that is, the force field inspection is passed), the ligand preparation (Ligand Preparation, see the red box on the upper right of the figure for the shortcut) is required.
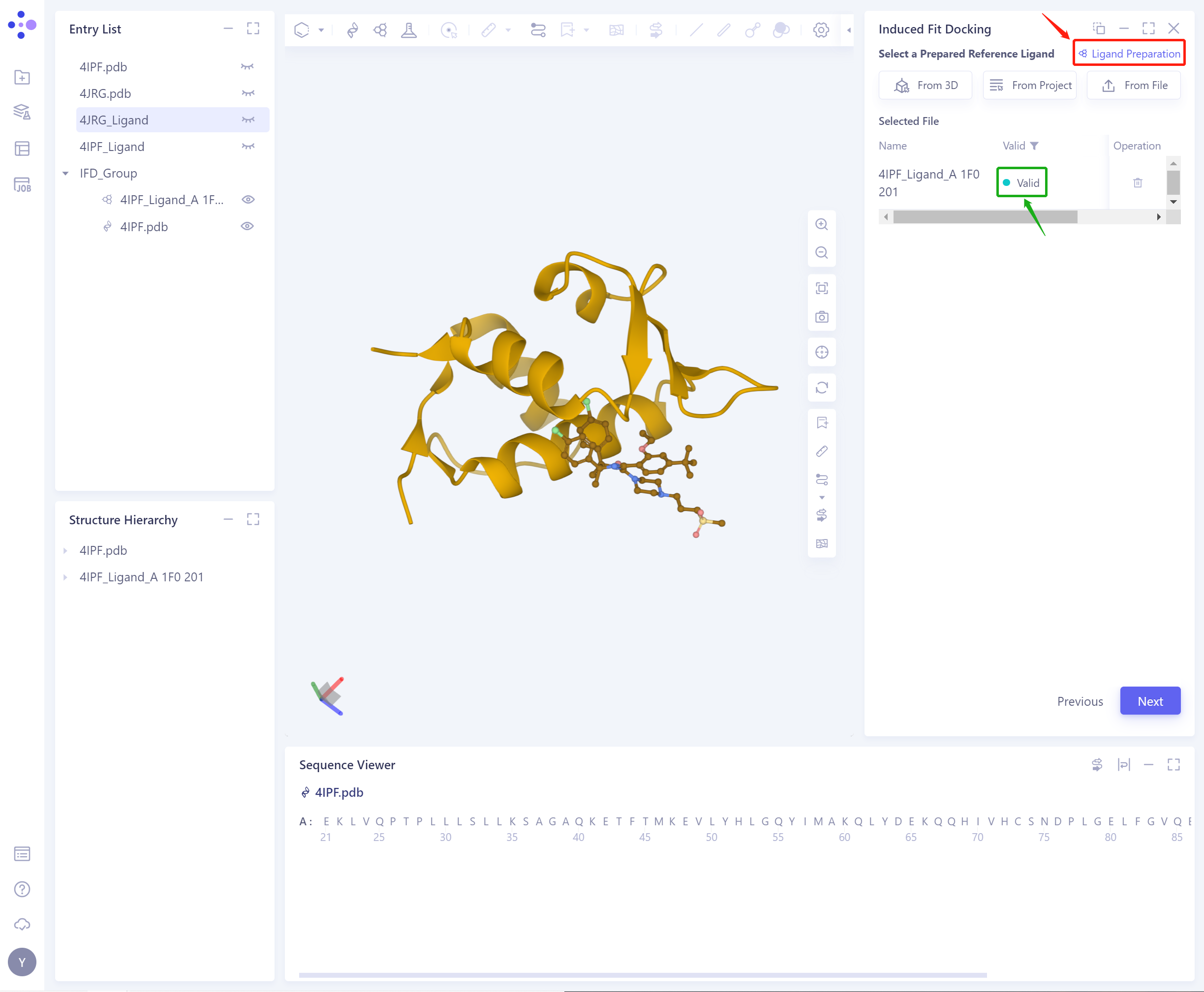
- After the ligand force field check is passed, click Next to continue to use the function.
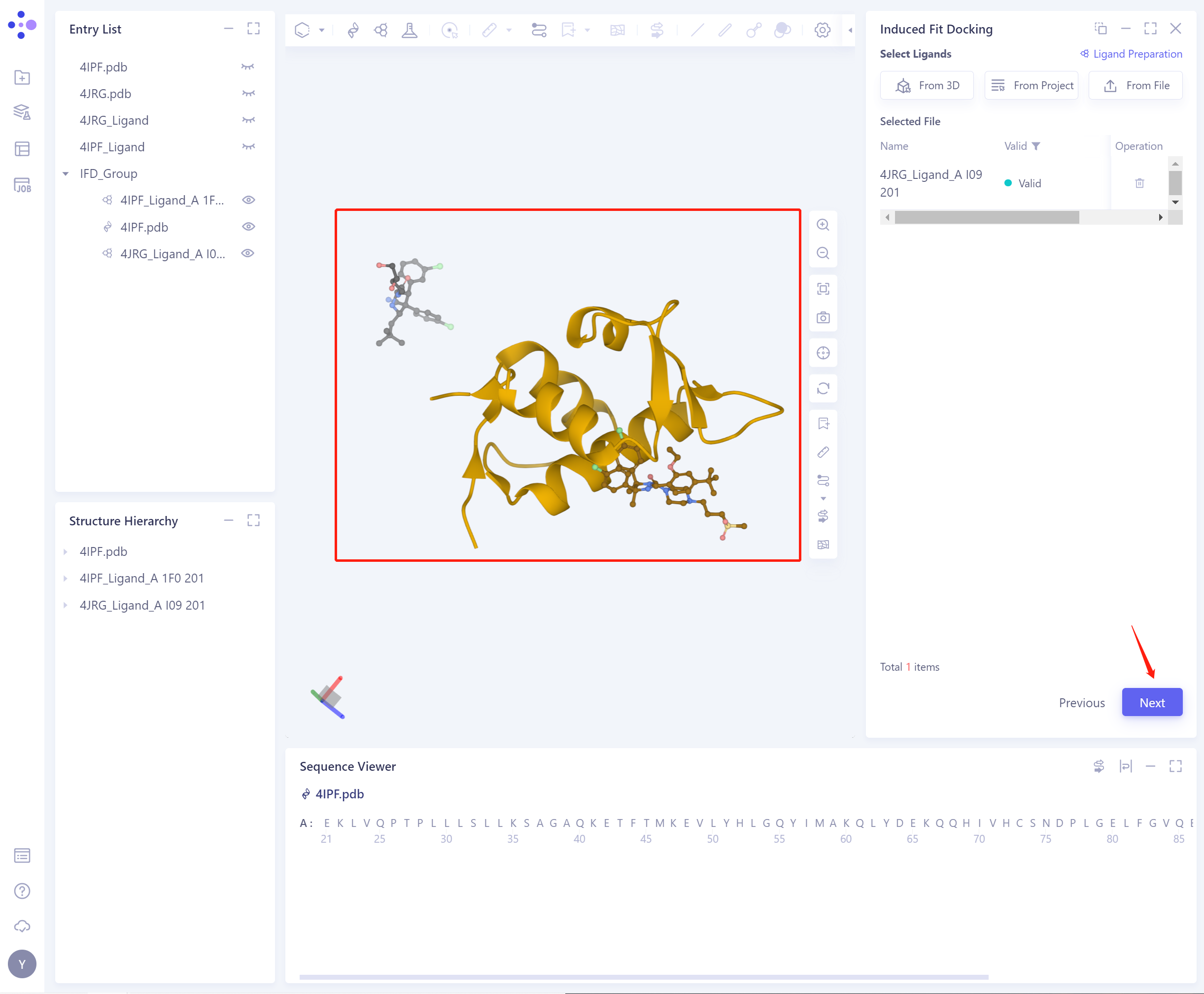
- Select Ligands: (There are three ways to import ligand, as above). The structures of the successfully imported protein and the two ligands are shown in the figure. Click Next to continue.
-
Pocket Setting:
-
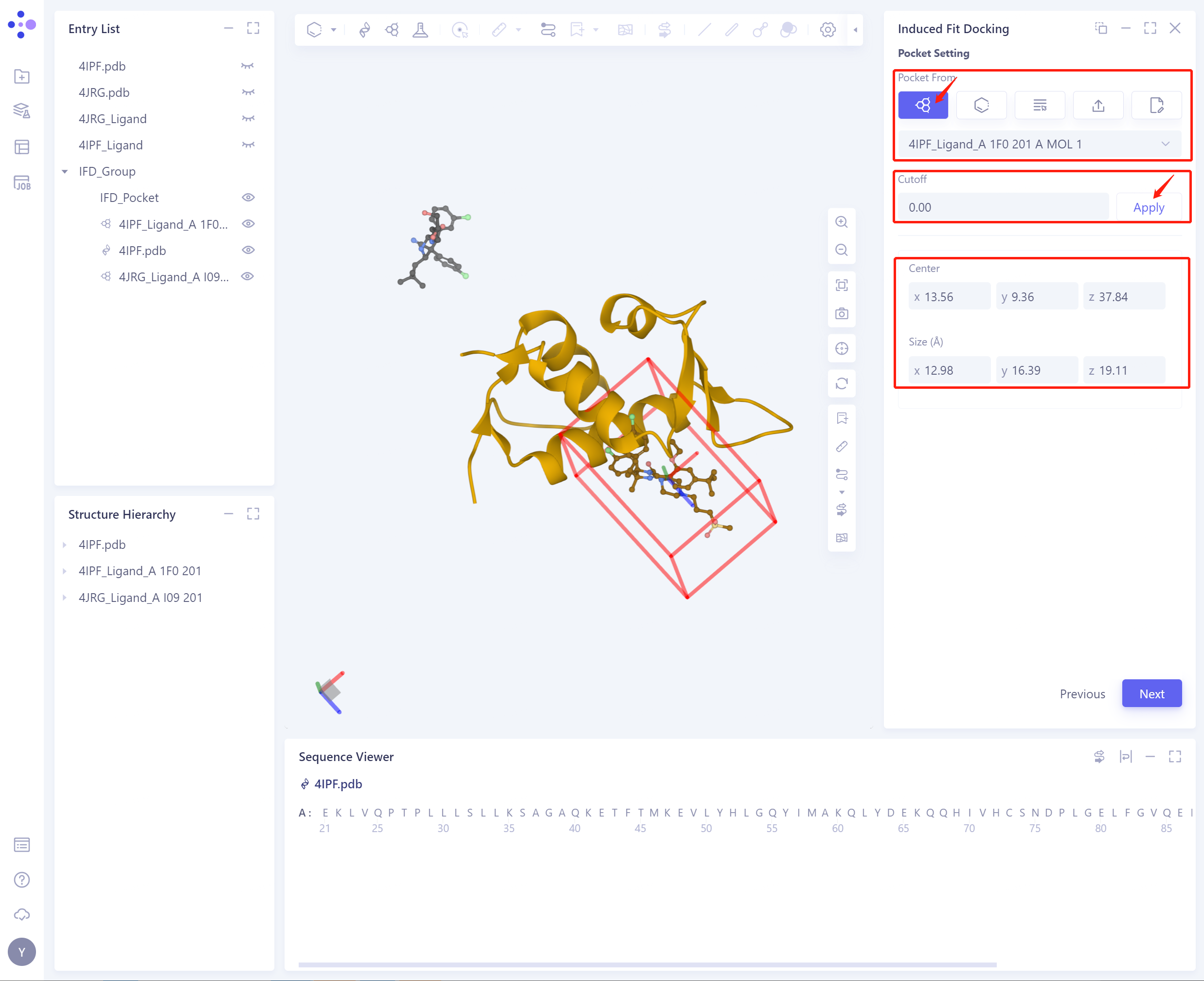
Pocket From: 5 pocket settings are supported:
-
1)Select Ligand in the structure as center: select Ligand in the protein as the center to determine the initial Pocket
-
Cut off: Set the Cutoff value, and the length, width and height of Pocket will be increased by 2 times of Cutoff;
-
Center: determine the center coordinate of Pocket according to the setting mode of Pocket;
-
Size (Å): Determine the volume of the Pocket based on the set center and cutoff (not less than 1000 1000 Å ³).
-
-
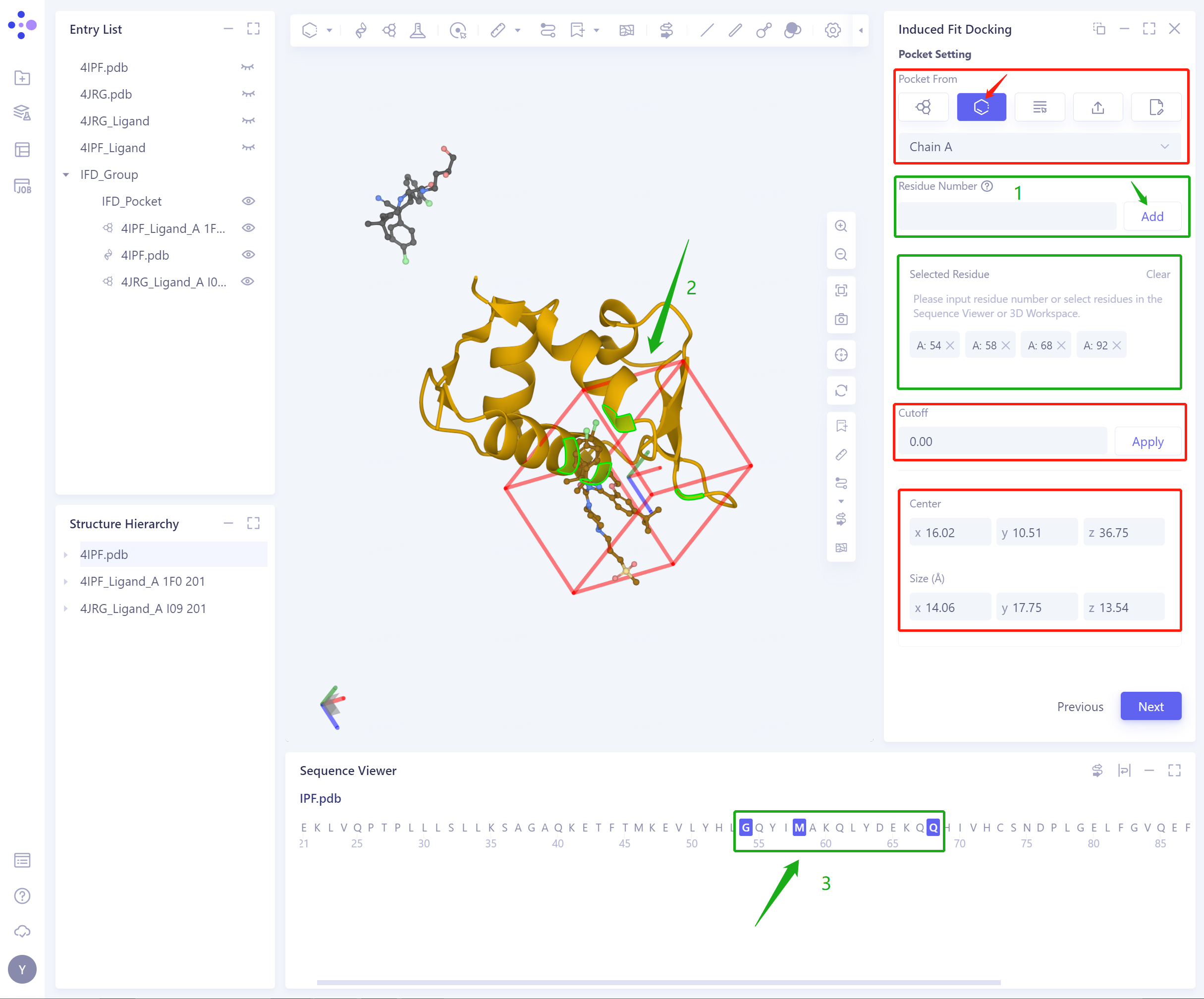
2)Select Residues as Center: selecte a protein residue as a center to determine an initial Pocket;
-
Residue Number: Set the Pocket position according to the residue number, support the input of Residue Number (1), and select residues in 3D workspace (2) and Sequence Viewer (3);
-
Cut off: Set the Cutoff value, and the length, width and height of Pocket will be increased by 2 times of Cutoff;
-
Center: determine the center coordinate of Pocket according to the setting mode of Pocket;
-
Size (Å): Determine the volume of the Pocket based on the set center and cutoff (not less than 1000 1000 Å ³).
-
-
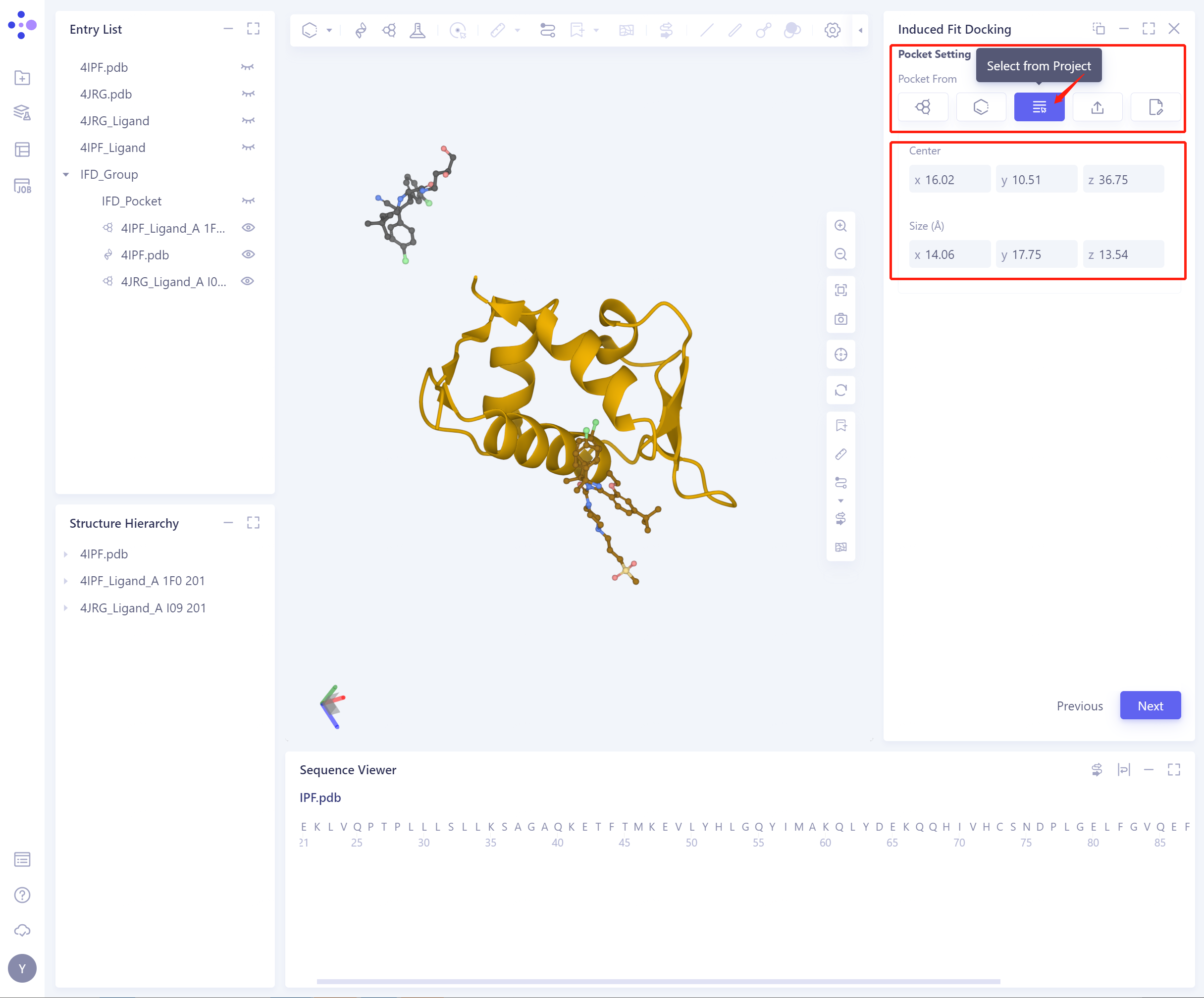
3)Select from project: Select the initial Pocket from the project.
-
Center: Support setting the center coordinates of Pocket;
-
Size (Å): Support setting pocket size to determine the volume of pocket (not less than 1000 1000 Å ³).
-
-
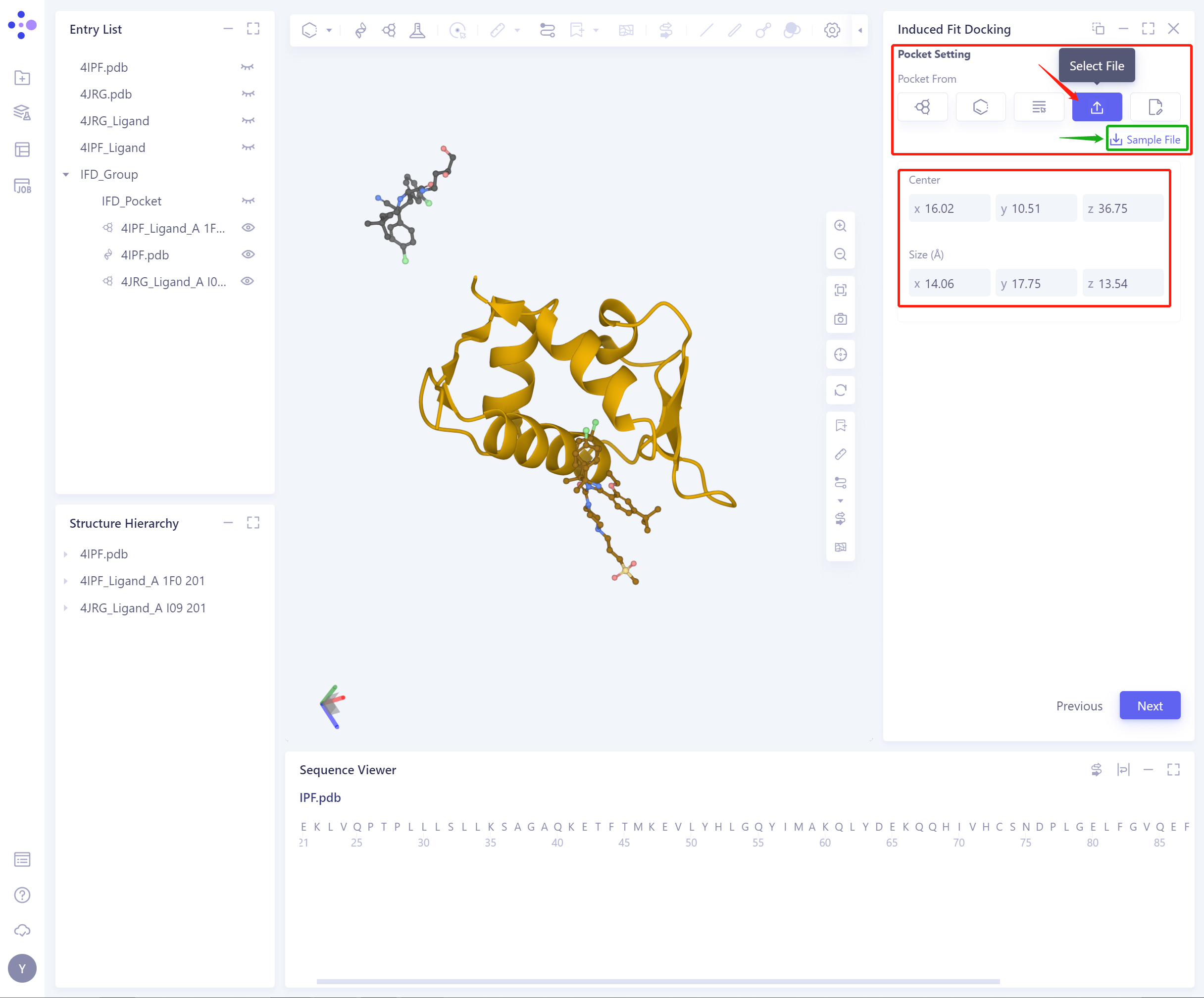
4)Select from File: Select from File to confirm the initial Pocket (Sample File that provides pocket settings)
-
Center: Support setting the center coordinates of Pocket;
-
Size (Å): Support setting pocket size to determine the volume of pocket (not less than 1000 1000 Å ³).
-
- 5)Input: directly and manually input Center coordinates and Size to set Pocket
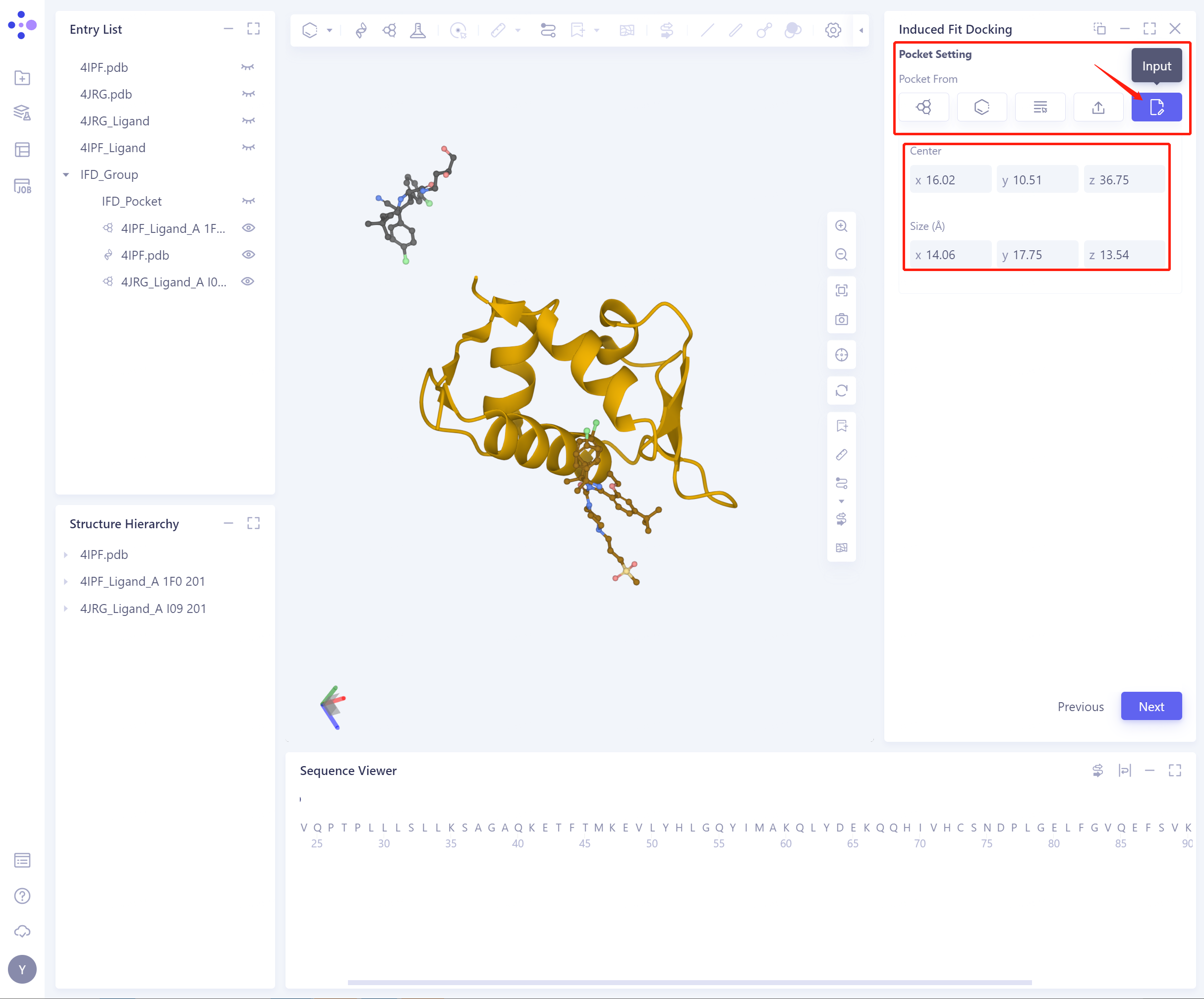
-
Select Refered Pharmacophore:
- Pharmacophore can be generated according to Referenece Ligand, including Hydrophobic, H-Bond Donor, H-Bond Acceptor, Cations, Cation, Anion;
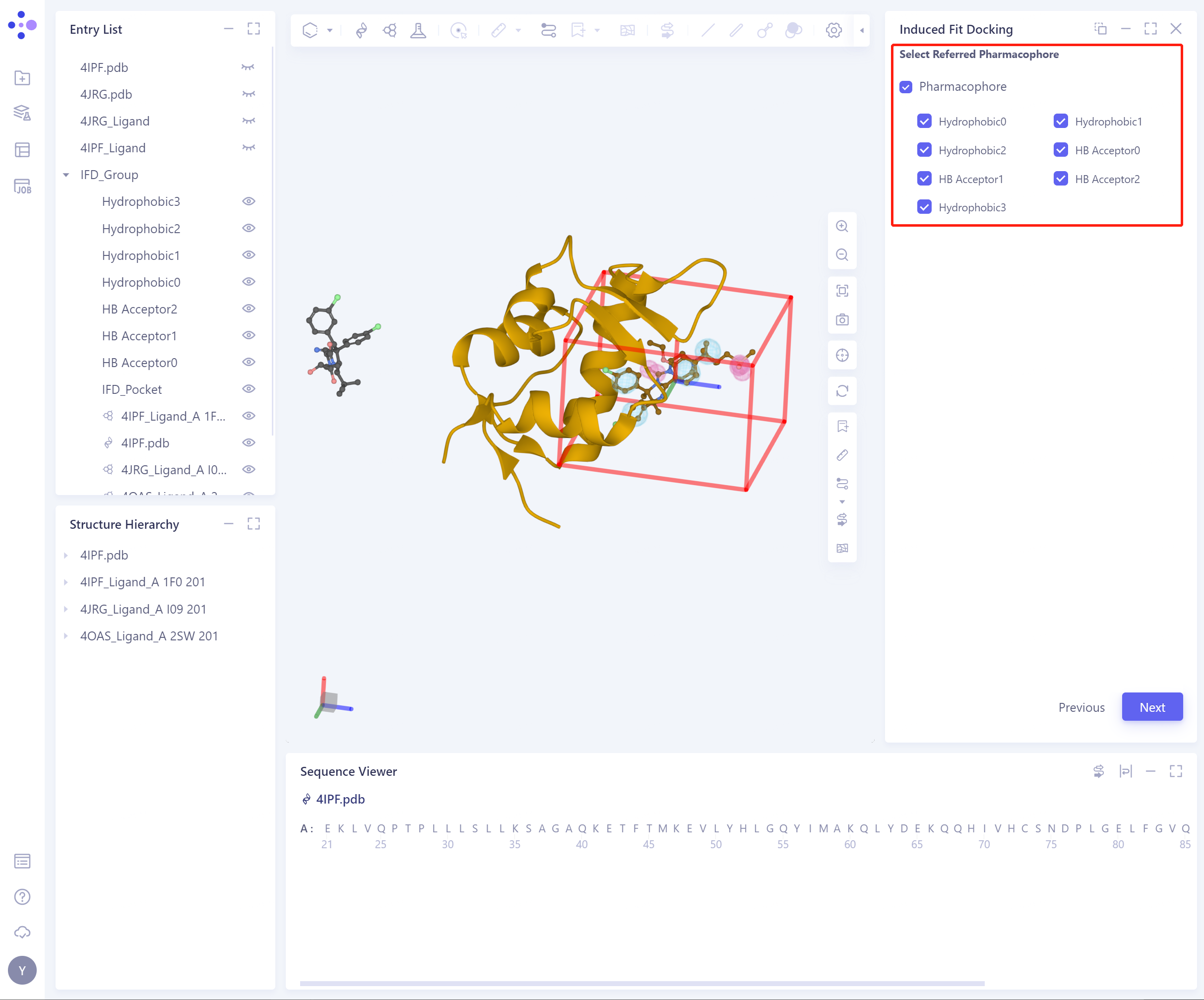
-
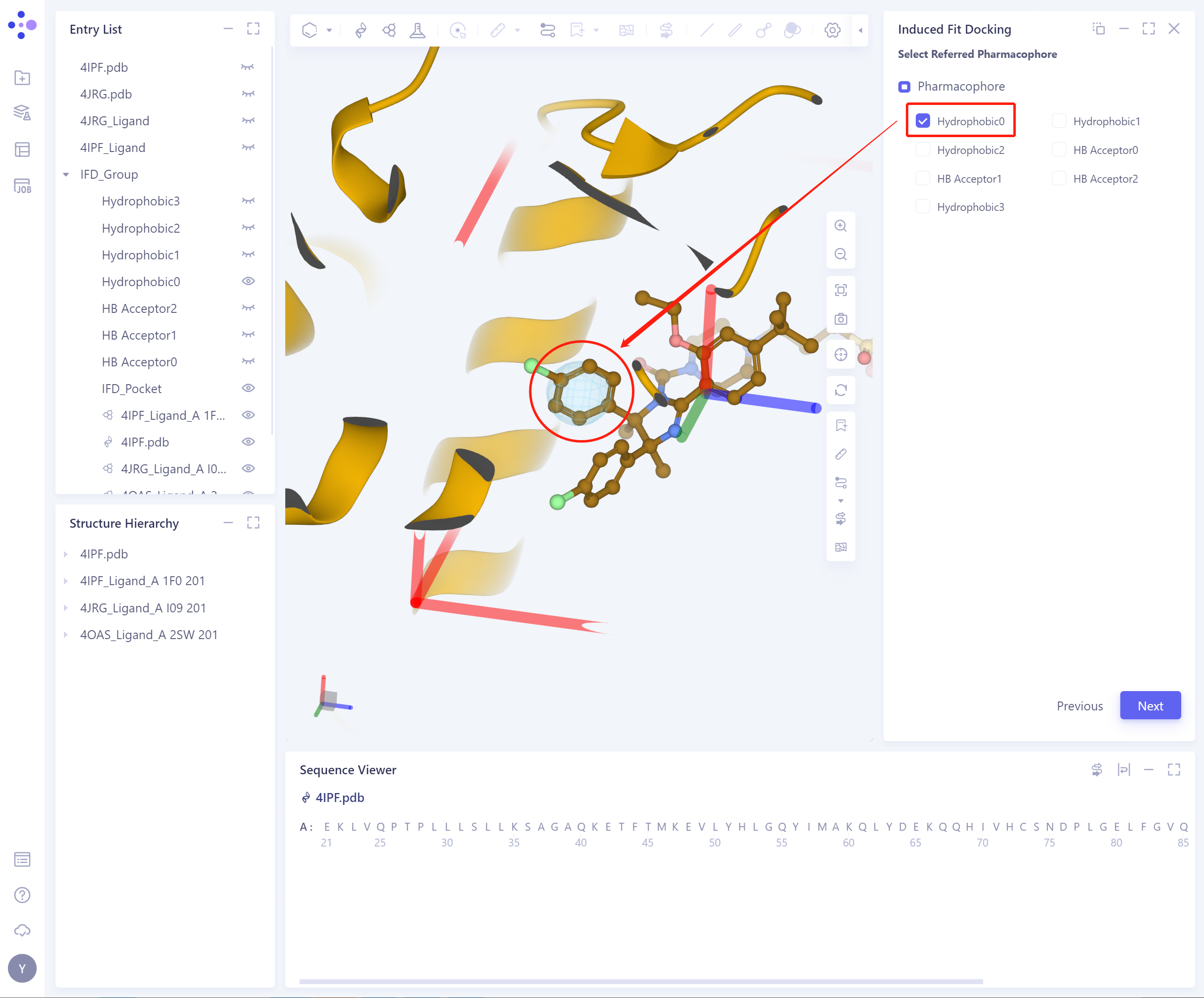
Check the corresponding pharmacophore, and 3D Workspace will automatically highlight the pharmacophore;
-
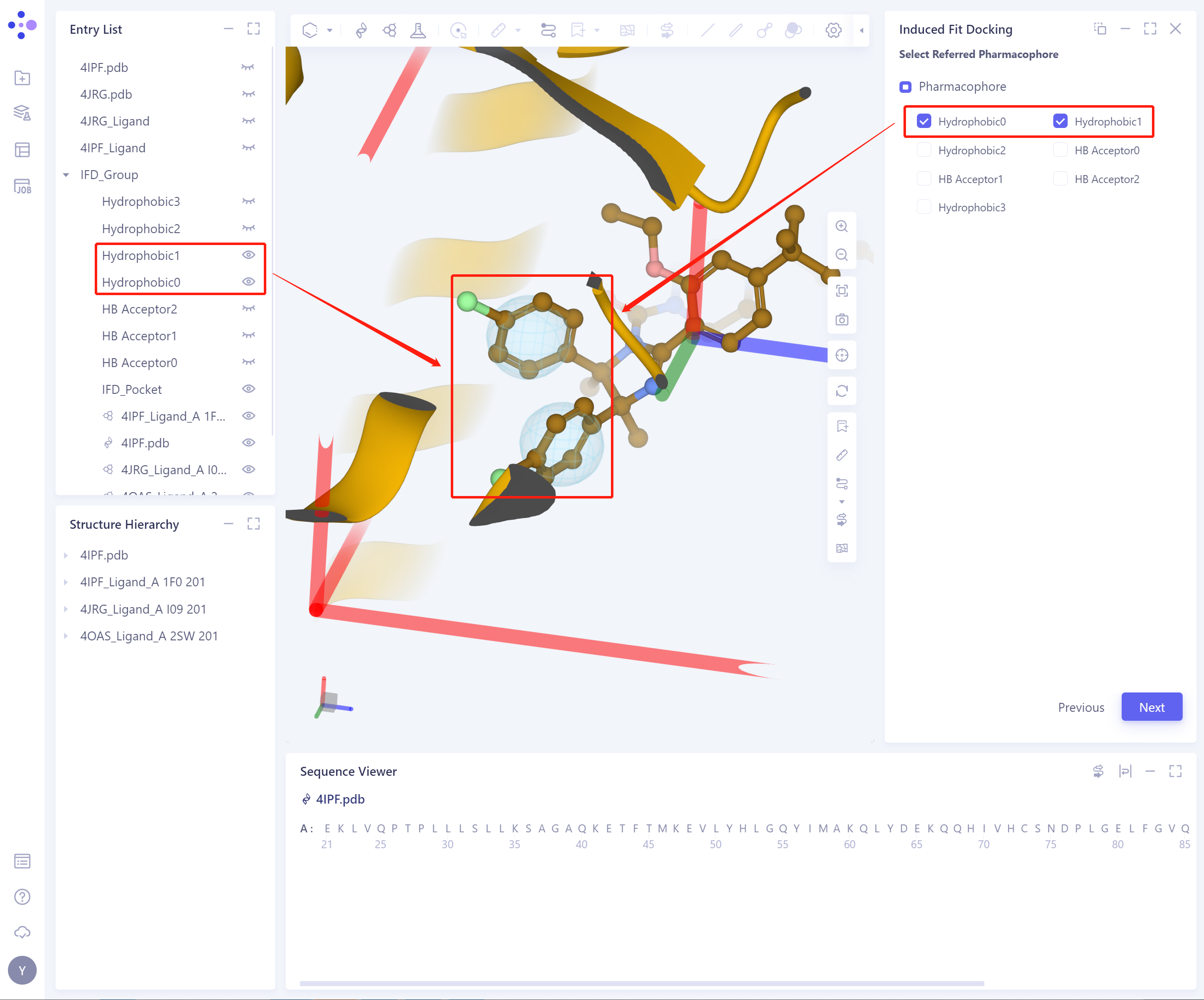
Depending on the pharmacophore chosen, the flexible residues of the protein can be determined.
- The appearance and concealment of pharmacophore can be directly controlled in Entry List.
-
Confirm and Setting: Confirm the input protein and ligand, and the selected pharmacophore is correct.
- Click Highlight All to highlight the selected pharmacophore.
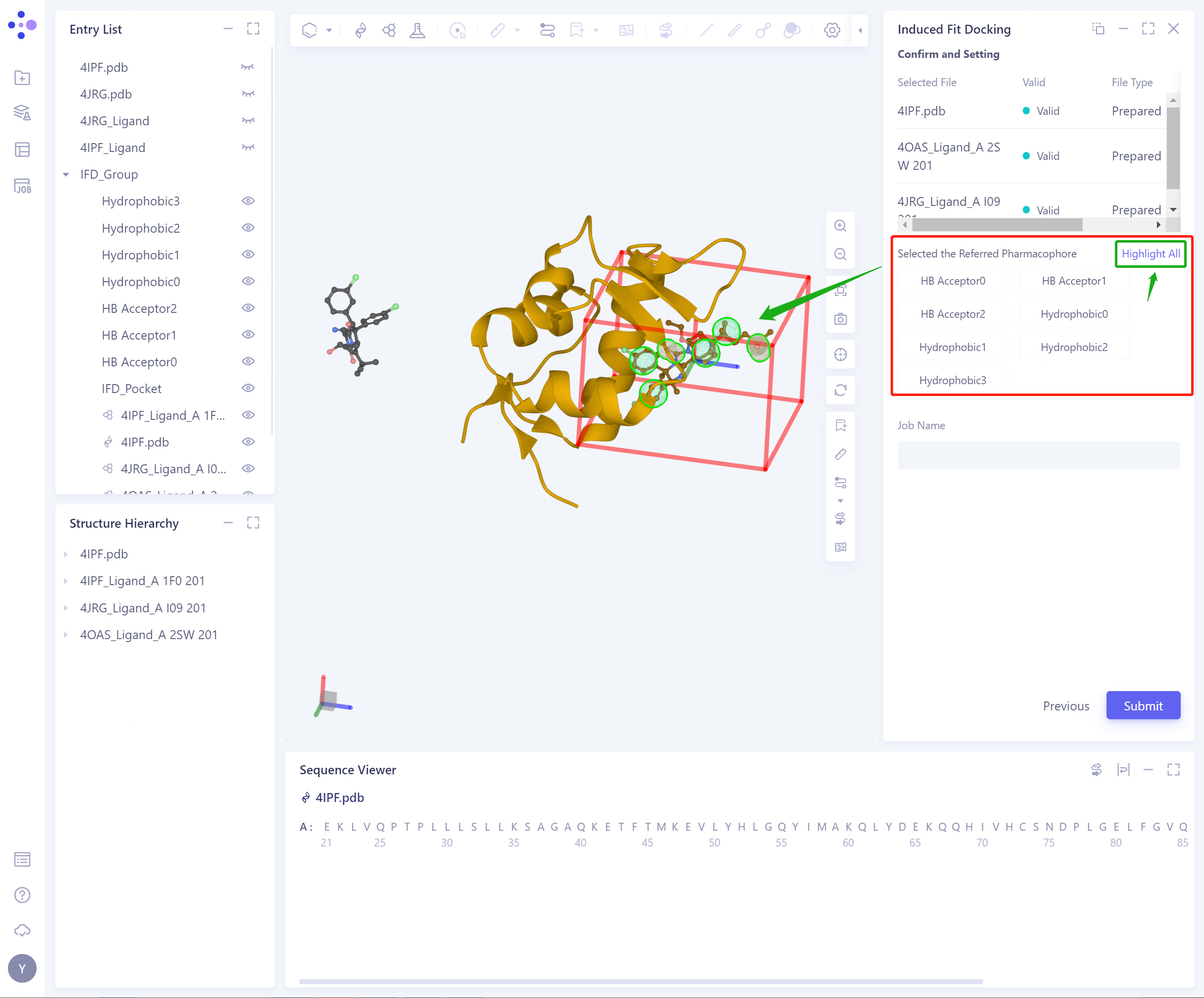
-
Job Name: Name the task.
-
Submit: Submit the task.
- Path 2: Whether to select Reference Ligand: Click No to unselect Reference Ligands
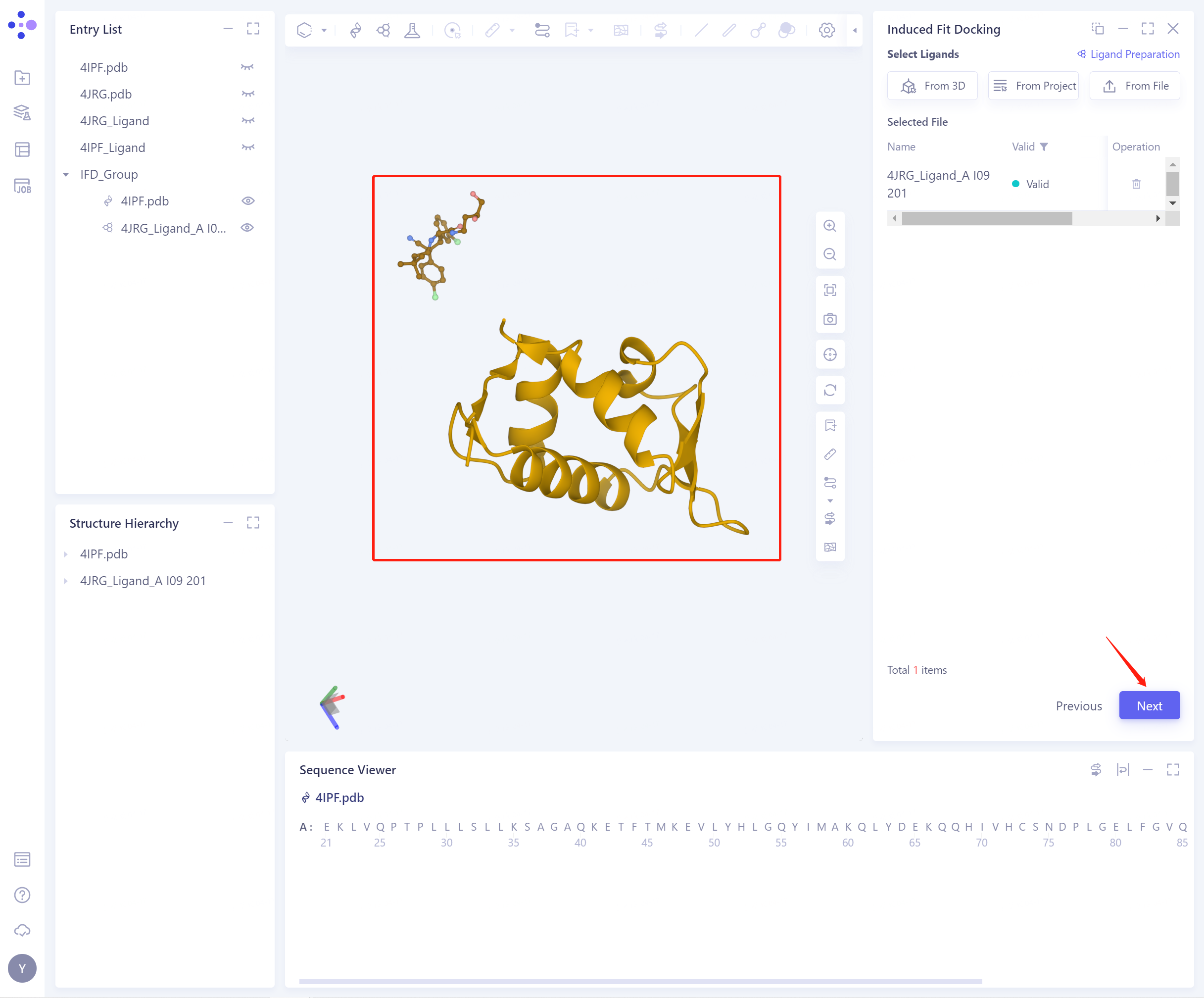
- Select Ligands (There are three ways to import ligand, and the specific operation is the same as above). The structure of the successfully imported protein and ligand is shown in the figure. Click Next to start Pocket Setting.
- Pocket Setting (There are 5 ways to set Pocket, and the specific operation is the same as above). The set Pocket is shown in the figure below:
-
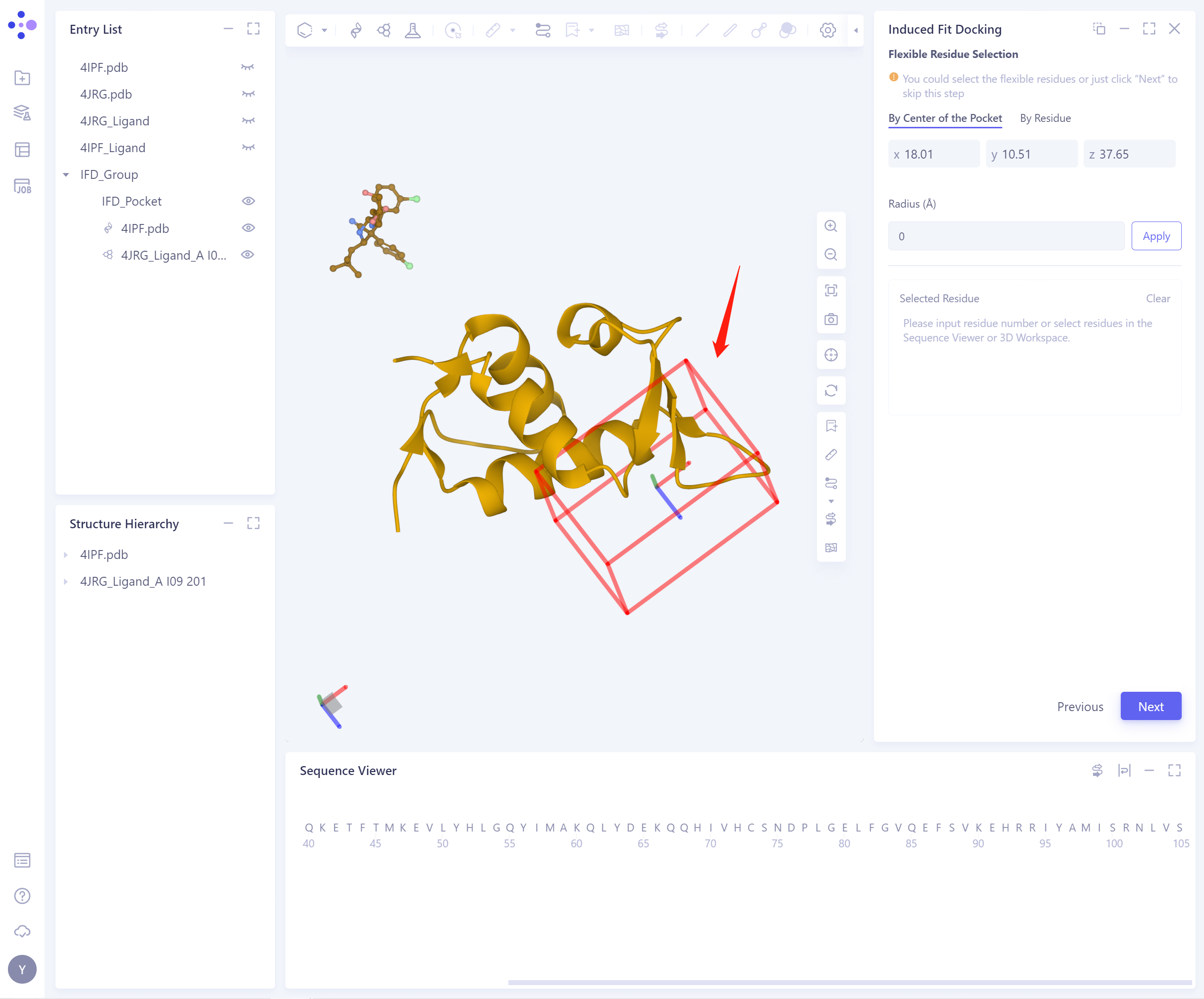
Flexible Residue Selection:
- By Center of the Pocket: select the extended range Radius according to the Pocket center, and determine the flexible residue (the selected flexible residue will be highlighted);
- By Residue: determine flexible residues by directly selecting Residue (a combination of 3D Workspace and Sequence Viewer is strongly recommended at this time);
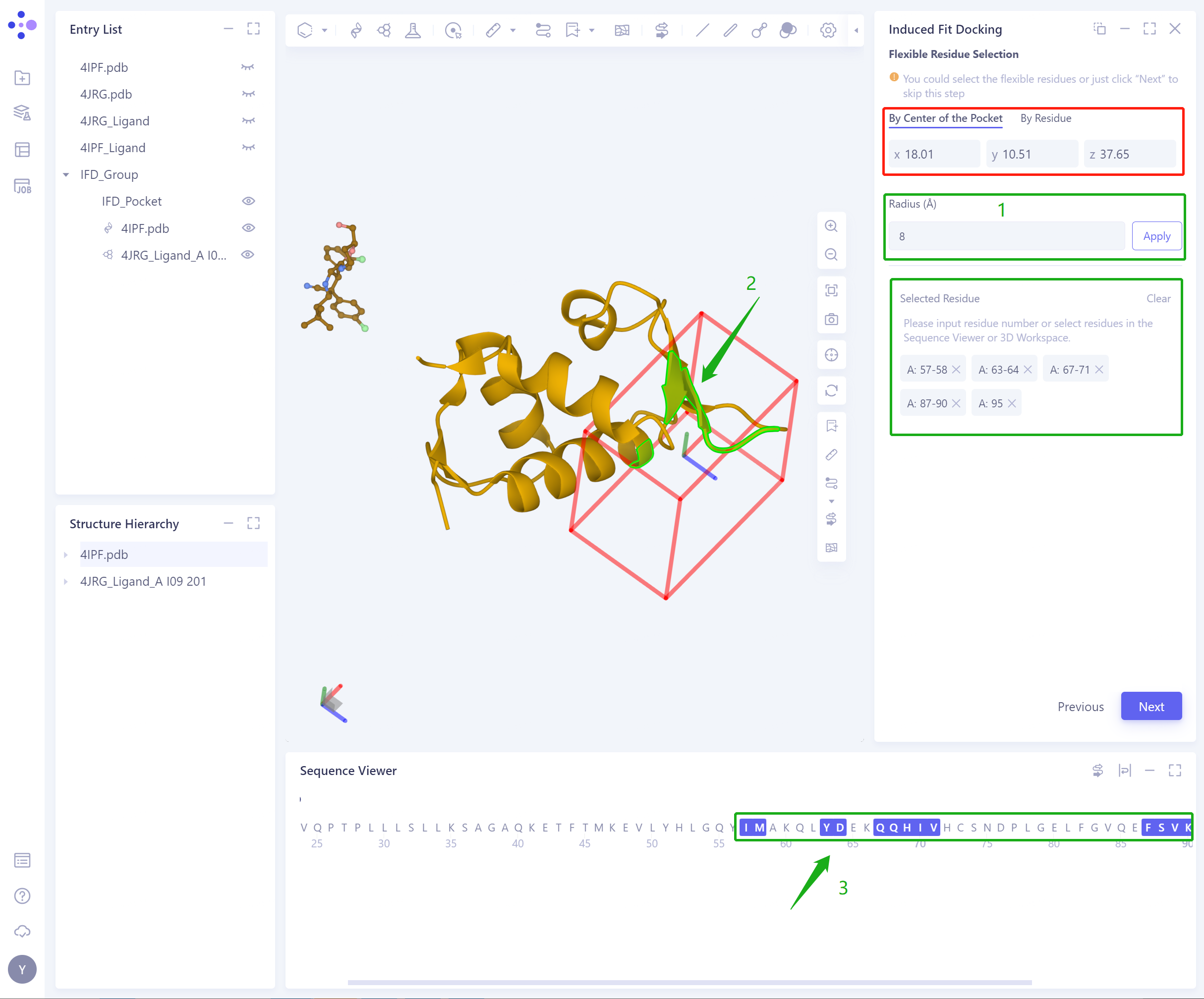
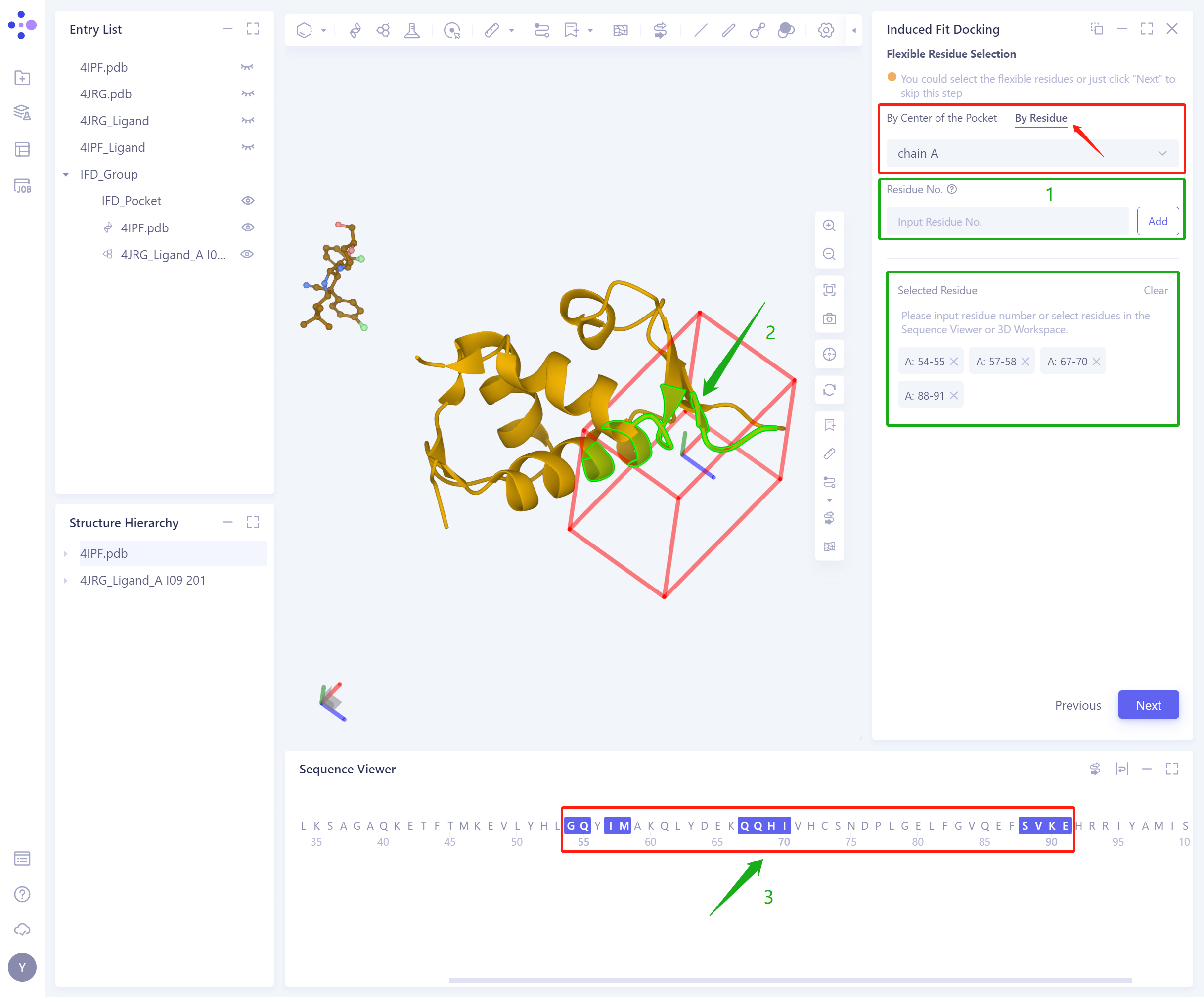
- The input protein and ligand were Confirm and Setting: determined, and the selected flexible residues were correct.
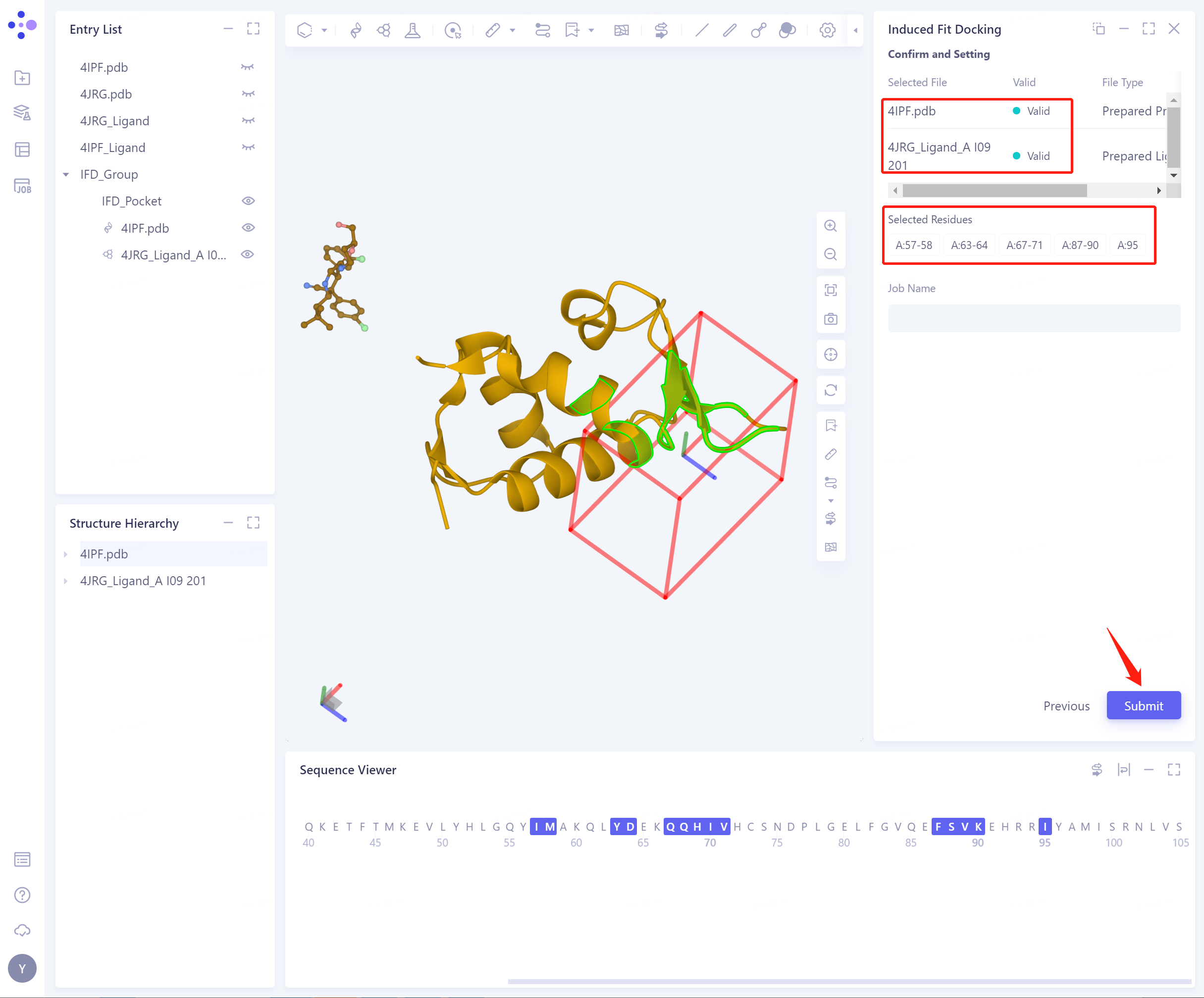
-
Job Name: Name the task.
-
Submit: Submit the task.
3. Analysis of results
3.1 Entrance
-
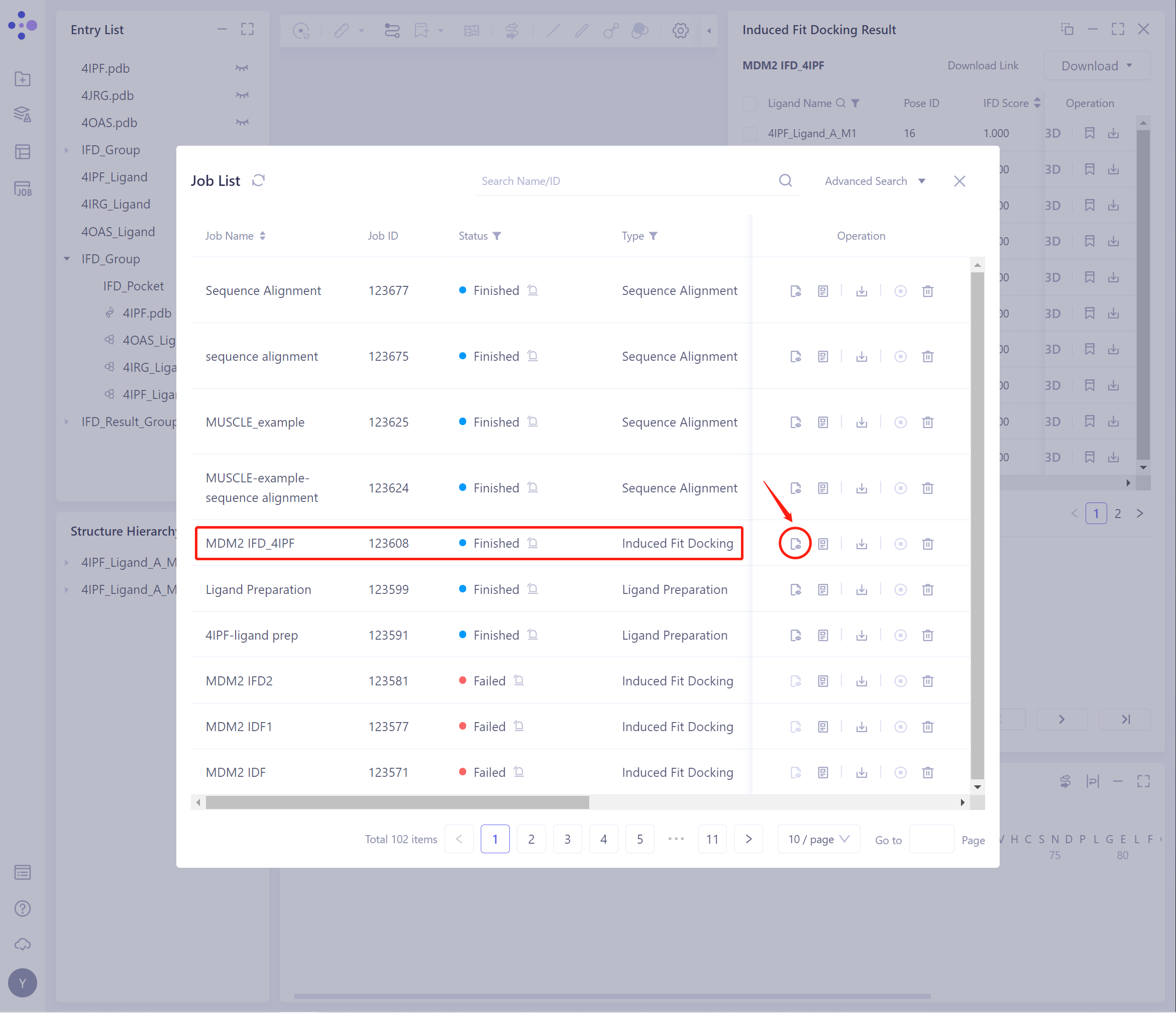
On the general menu bar on the left, Job → find the Induced Fit Docking Job.
- The task can be found by searching for the Job Name, or by filtering the Job Type.
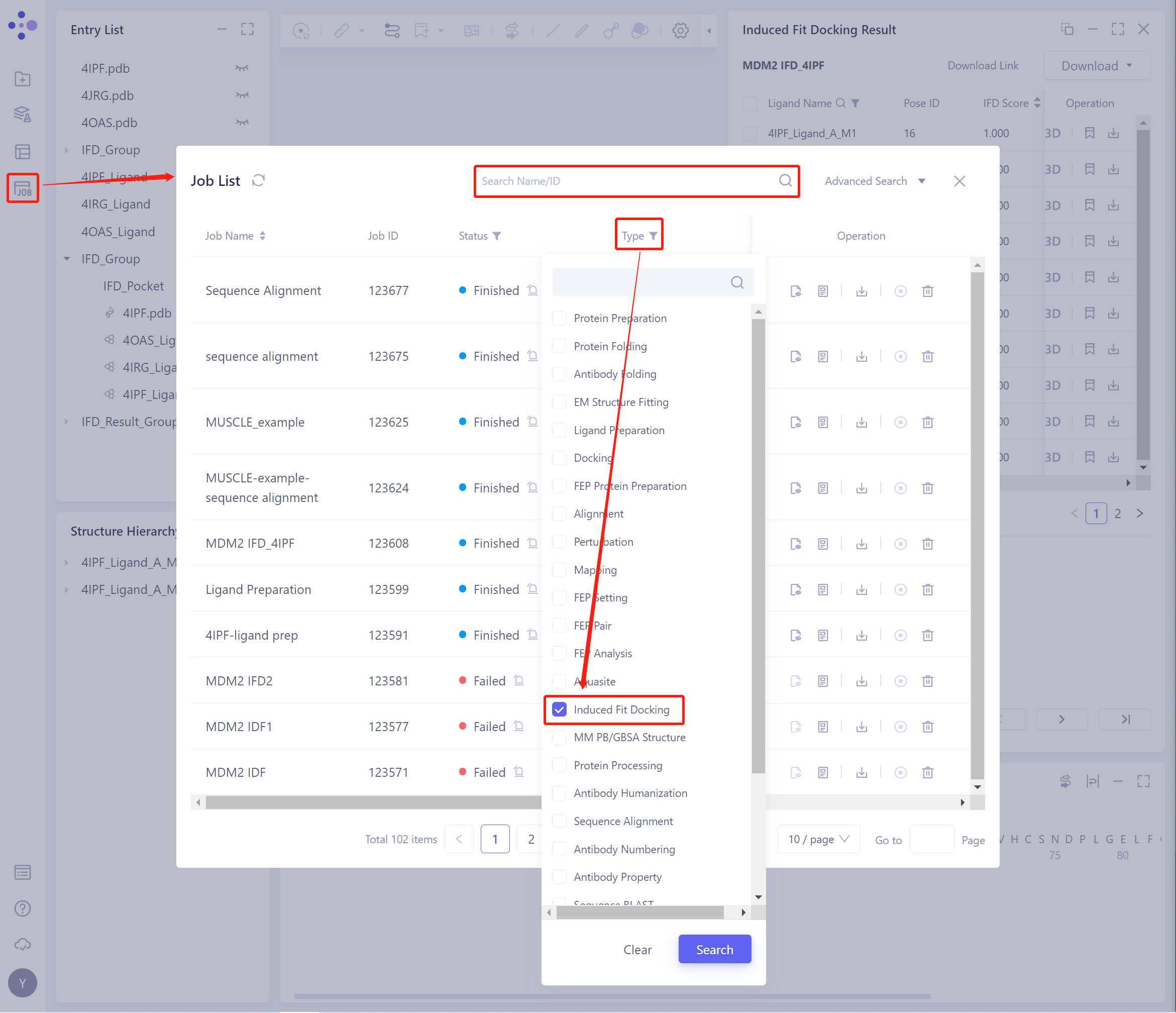
- Select the task to be viewed, and click Show in the Operation column to display the result of the task.
3.2 Results presentation
-
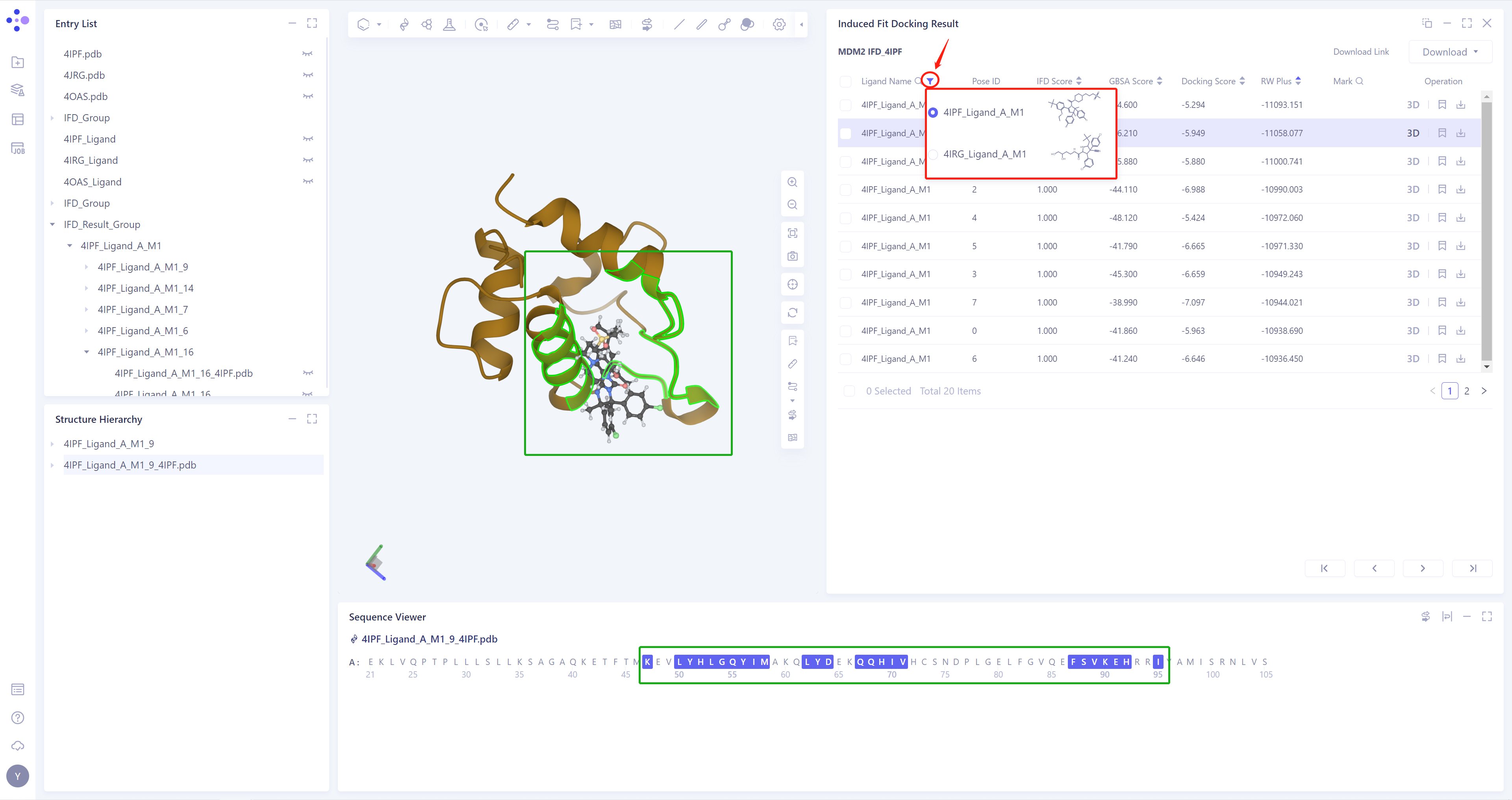
The results of the Induced Fit Docking will be automatically displayed in the 3D Workspace, and the flexible residues under the corresponding Ligand conformation will be highlighted (linked display in the Sequence Viewer), as shown in the figure;
-
If multiple ligands are input, click the button in the red box to switch the ligand results.
-
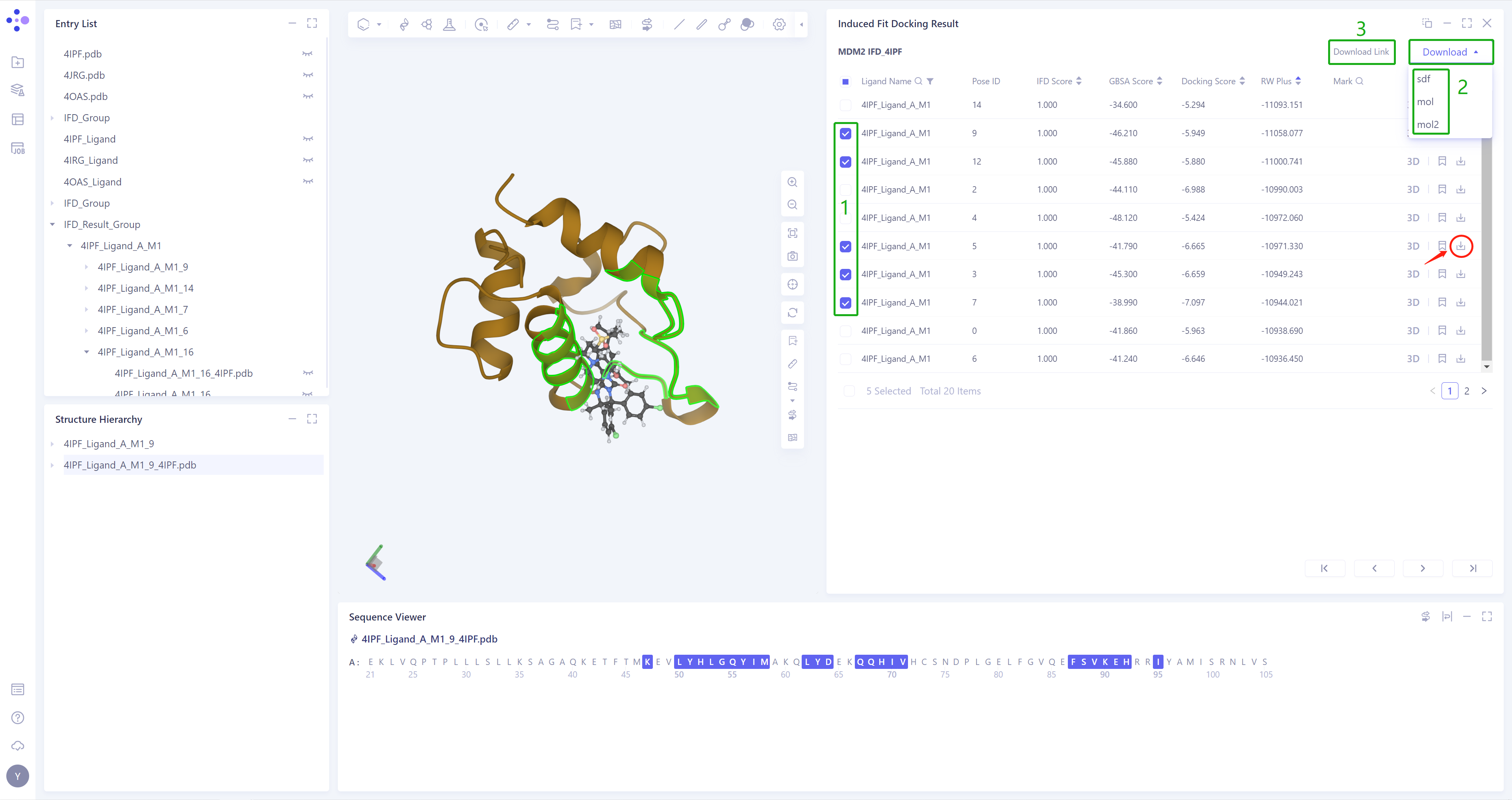
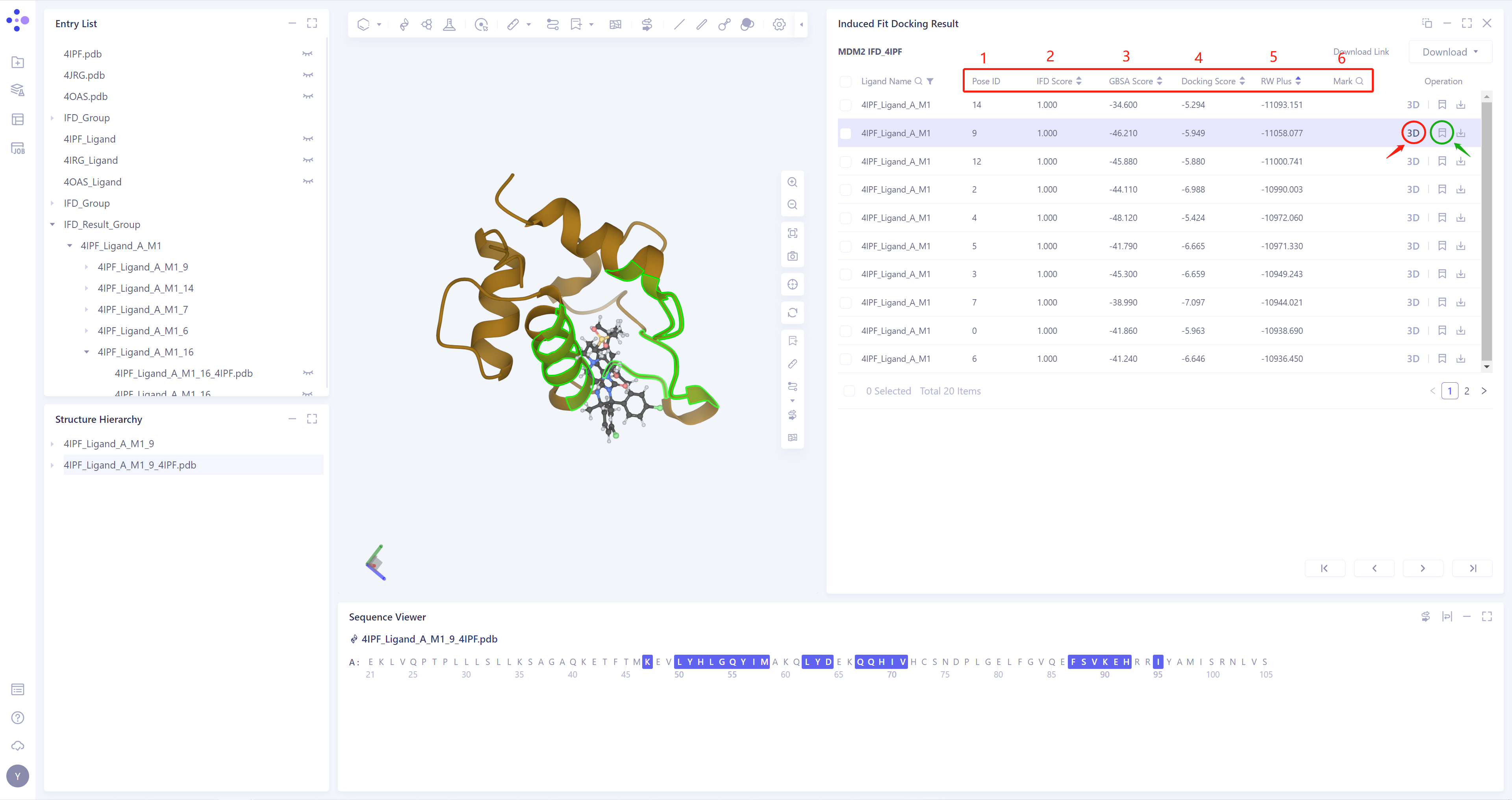
Provides five parameters for Docking results:
-
1)Pose ID: sort the Docking Pose according to the scoring function;
-
2)IFD Score: According to the binding mode of protein and ligand, give a comprehensive score (between 0 and 1);
-
3)GBSA Score: According to the binding mode of protein and ligand, the GBSA model gives a score;
-
4)Docking Score: the score given by Vina according to the binding mode of protein and ligand;
-
5)RW Plus: RW Plus scoring function (can handle scoring items containing protein side chains);
-
6)Mark: For some special data lines, you can click mark (in the green box) to make manual remarks, and search by remarks is supported;
-
-
Click 3D (red box) in the Operation column to display the result of the corresponding pose in the 3D workspace, and support the keyboard ↑ ↓ key to switch the pose (Note: this operation is switched according to the order of Pose ID);
-
The result of each Pose can be downloaded (red box), and batch download of multiple Pose results is supported;
-
As there are many files to be packed and downloaded after checking, you can click Download, and then click Download Link to get the download link;
-
Support downloading.sdf/.mol/.mol2 format files.