Binding Stability
1. Introduction
The process of binding small molecule drugs to proteins is called Binding. Small molecules are stably bound to proteins with a certain Binding Pose. Understanding the Binding Pose of protein- ligand complexes at the atomic level can provide us with ideas for drug design in the optimization stage of lead compounds for drug development. Therefore, it is particularly important to use molecular dynamics (MD) to verify the stability of small molecule Binding Pose.
The Hermite platform's Binding Stability module provides a molecular dynamics simulation-based method for measuring the stability of small molecule Binding Pose in small molecule-protein complexes.
2. Usage
2.1 Entrance
Left Common Menu Bar Function → Virtual Screening → Binding Stability.
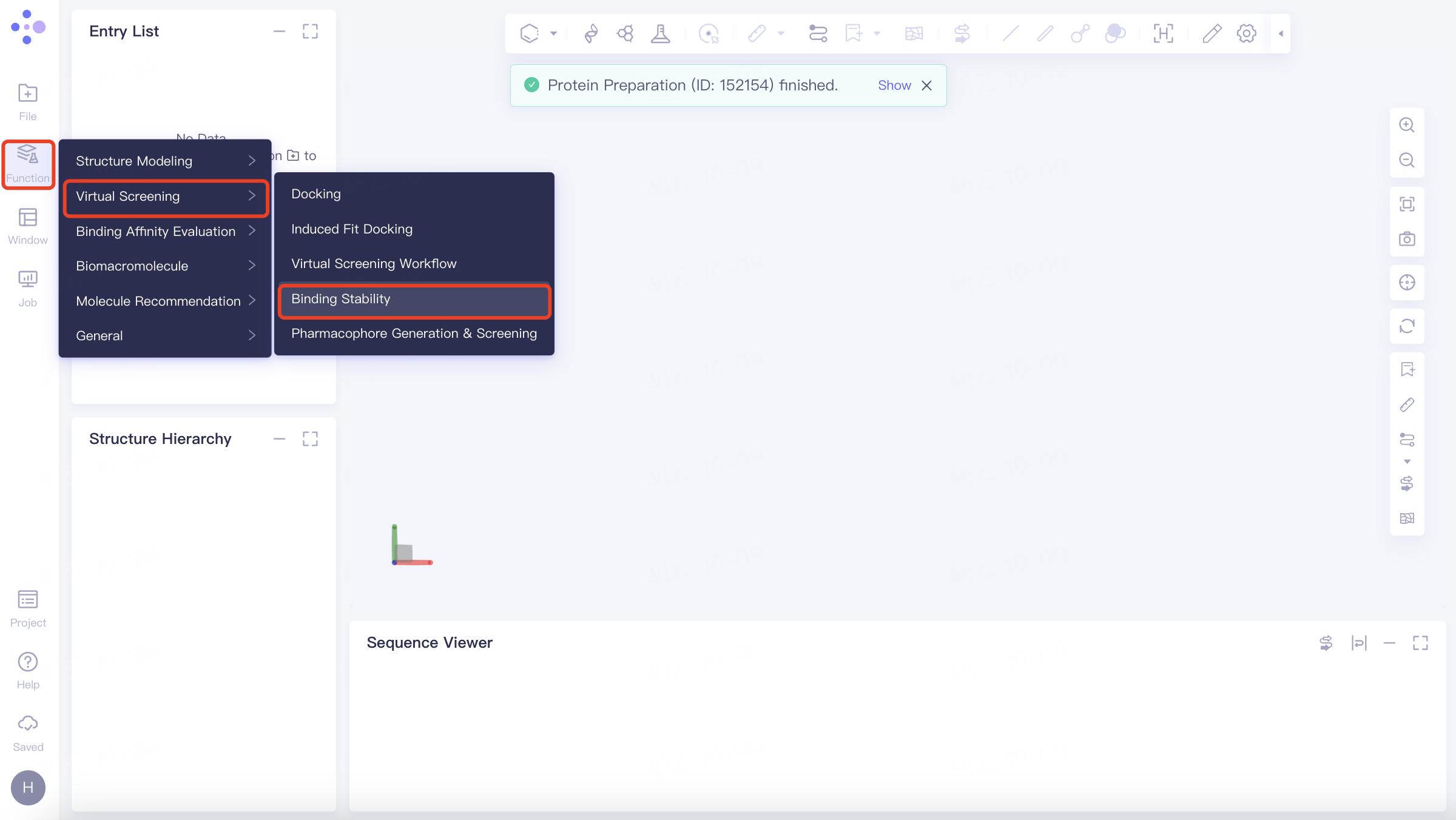
The Binding Stability operation box (shown in red) appears on the right side, and the overall interface is as follows:
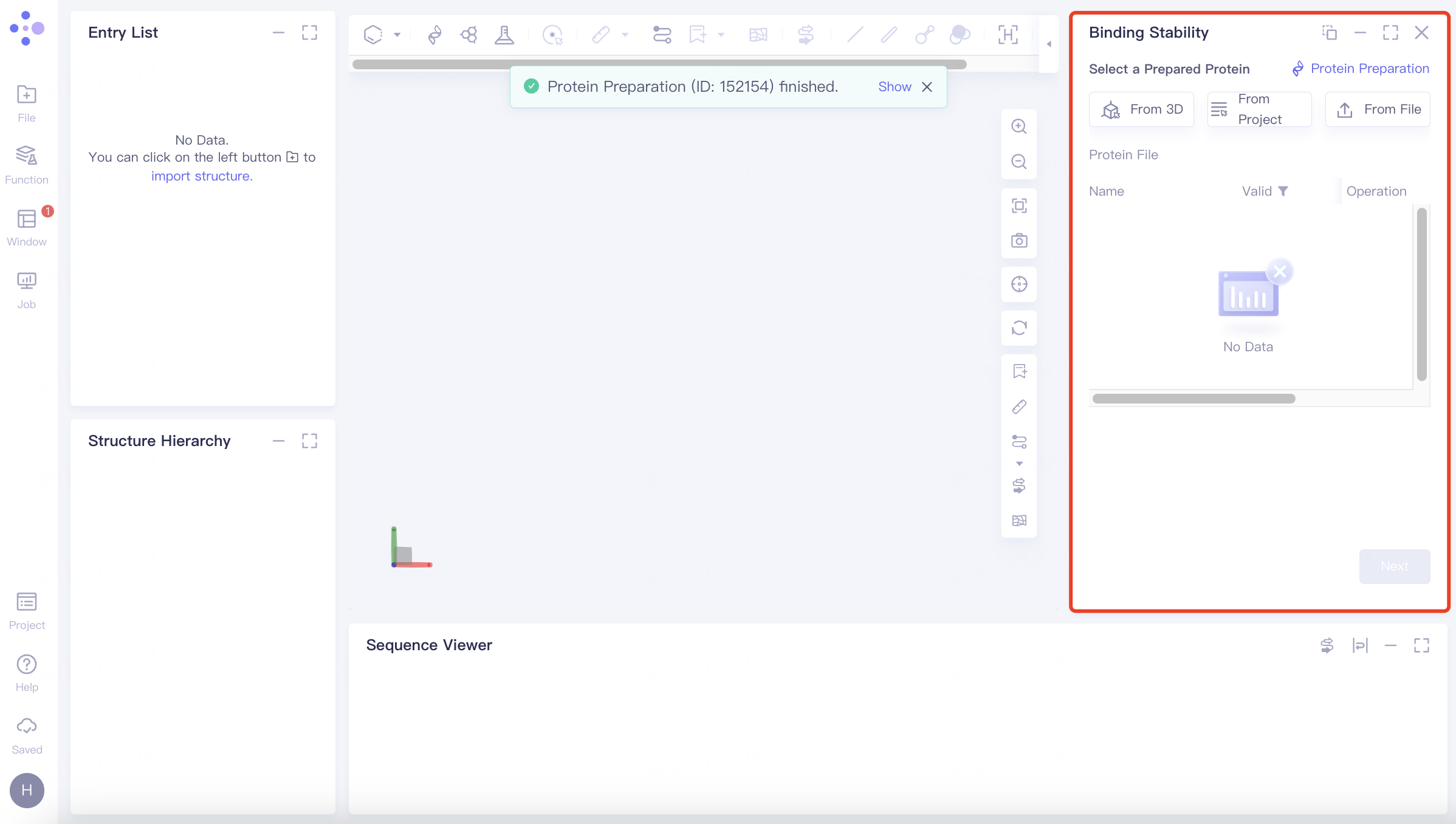
2.2 Operation
2.2.1 selective protein
Upload protein structures in the following three ways
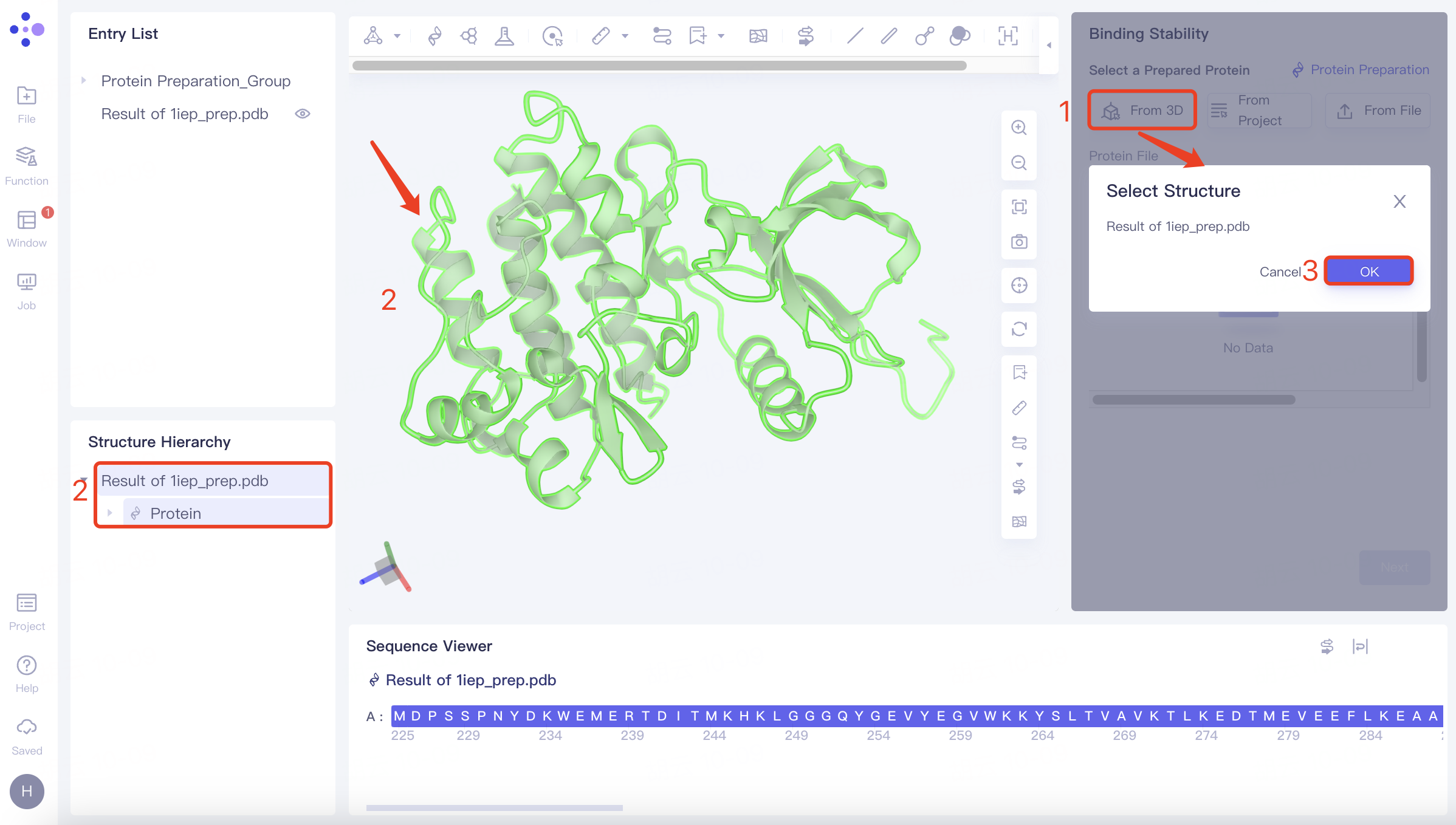
(1)From 3D:
Click the Select from 3D Workspace checkbox → Pop up the Select Structure interface → Select the desired protein structure in the Structure Hierarchy box on the left side of the interface/Select the protein structure in the 3D Workspace window → Select Structure interface to display the selected protein name, click OK.
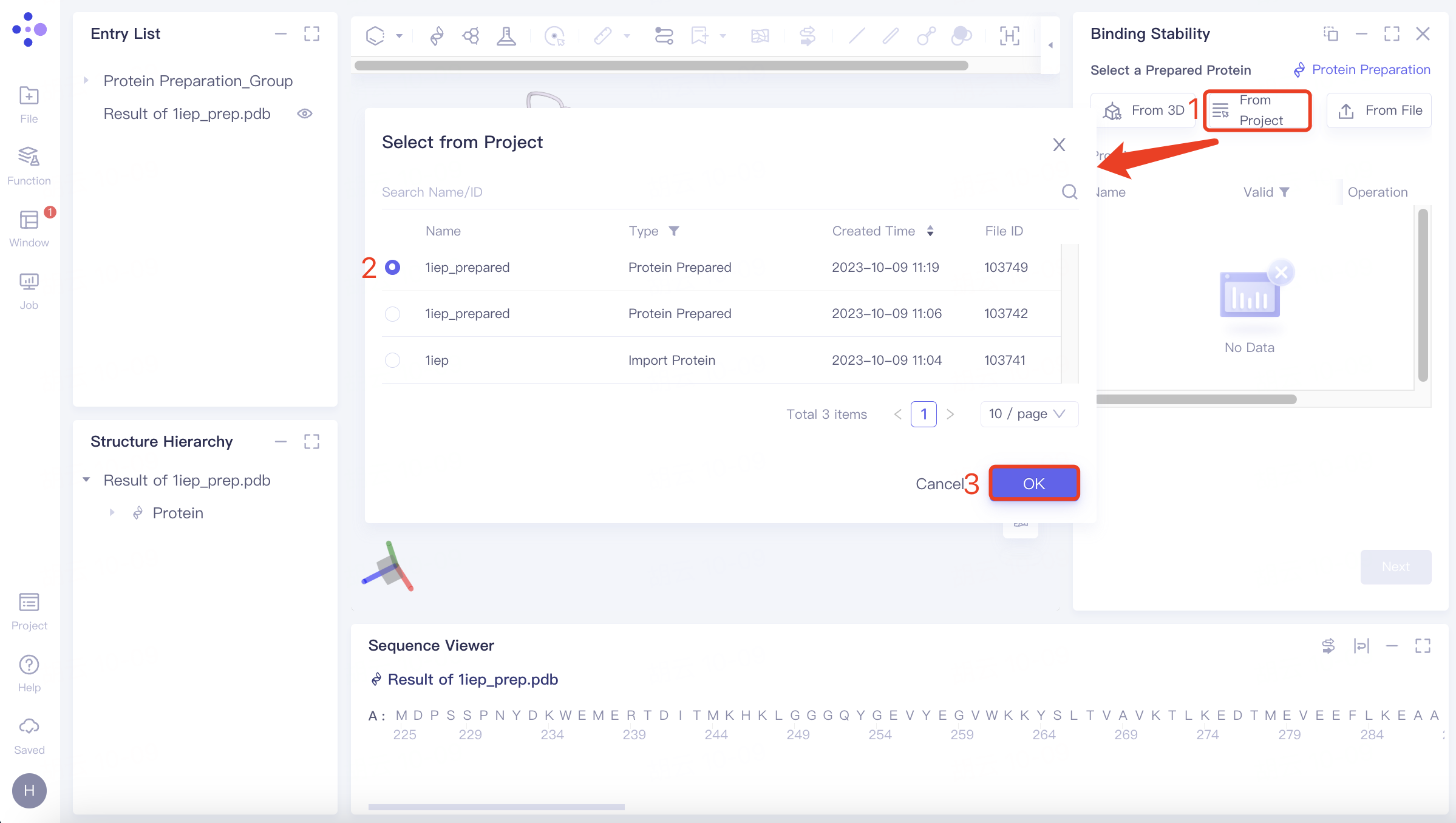
(2)From Project:
Click the “Select from Project” checkbox → pop up the “Select from Project” interface → select the desired protein structure → click “OK”.
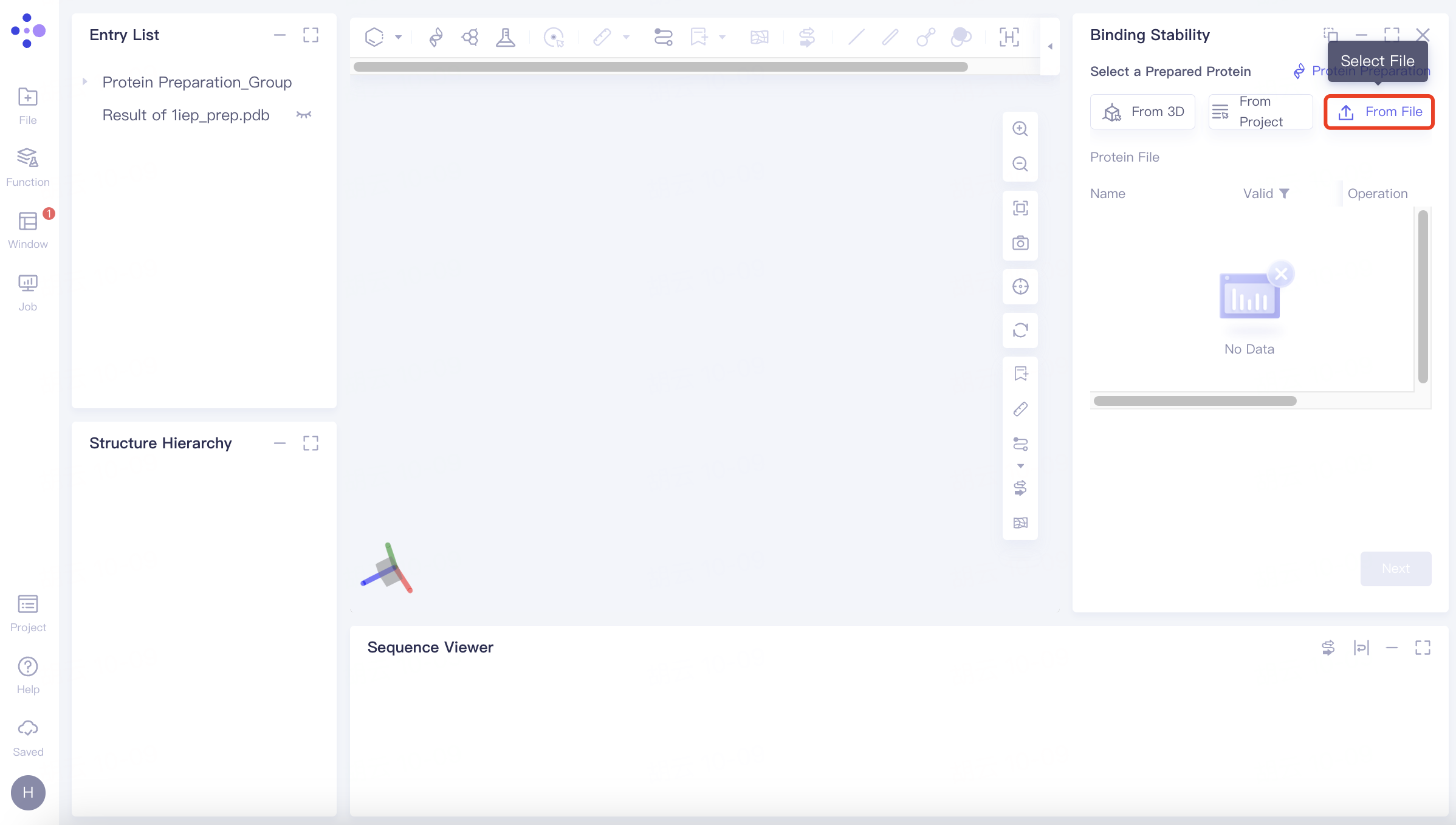
(3)From File:
Click the Select File checkbox → Select the desired protein file (.pdb format file supported) from the local folder and upload it.
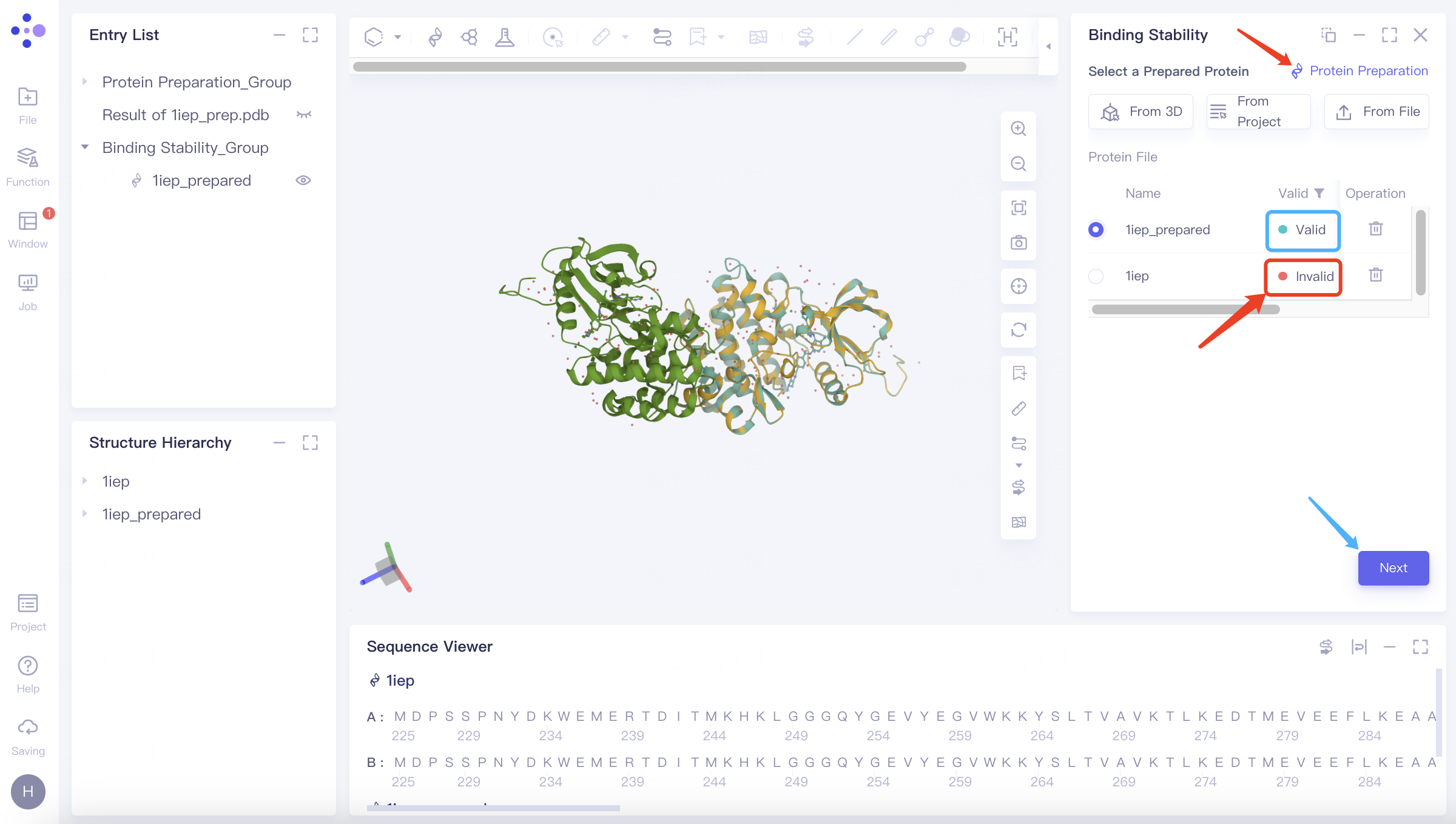
After passing the protein test, click Next to proceed to the next step.
When the protein test does not pass, it will display "Invalid". You can prepare the protein through "Protein Preparation" in the upper right corner. When its status is displayed as "Valid", it means the test has passed.
2.2.2 Upload Reference Ligand
There are three ways:
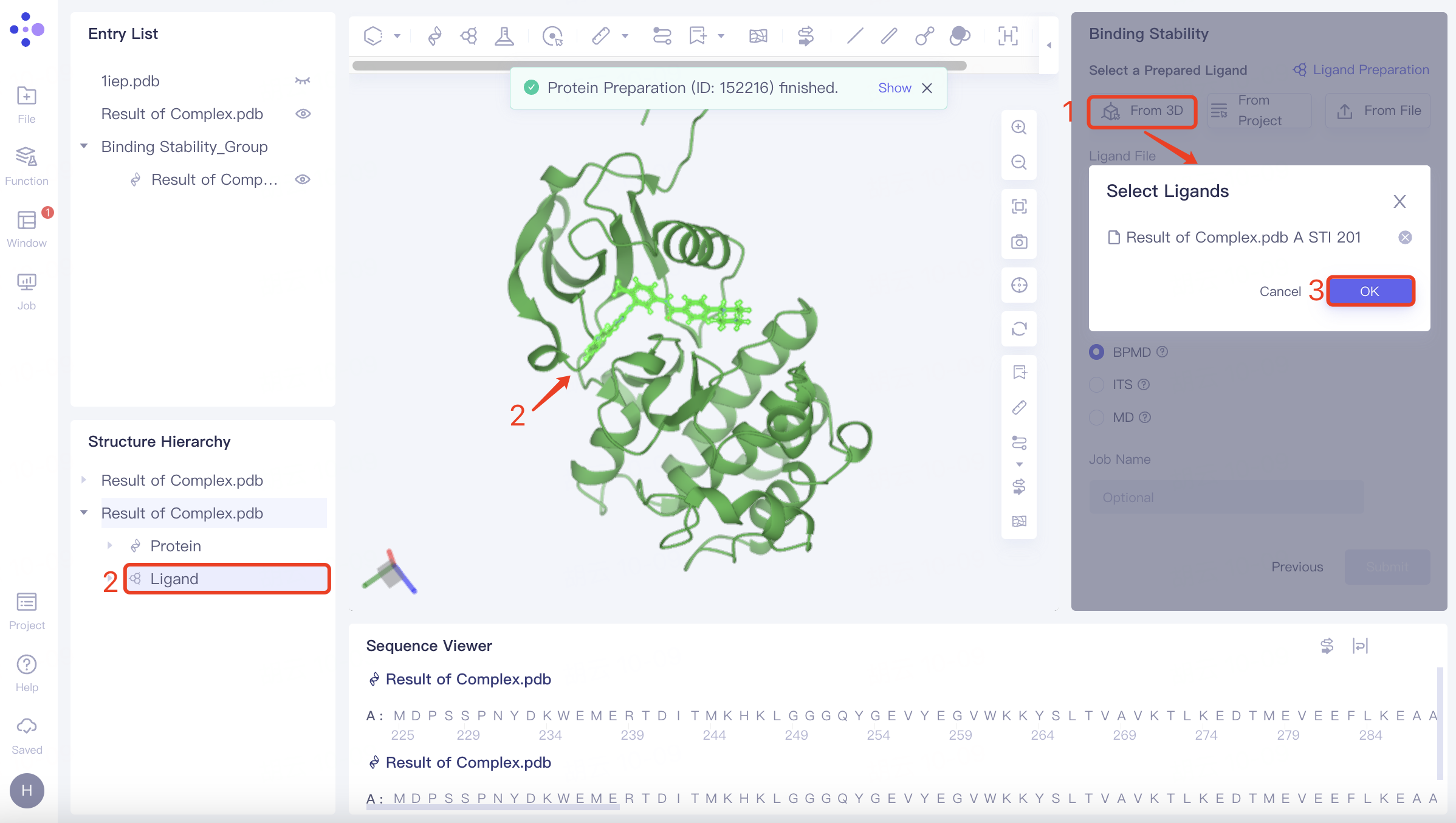
(1)Select from 3D
Click Select from 3D checkbox → Pop up Select Ligands interface → Select the desired ligand structure in the Structure Hierarchy box on the left side of the interface/Select the ligand structure in the 3D Workspace window → Select Ligands interface displays the selected ligand name, click OK.
(2)Select from Project
Click the Select from Project checkbox to pop up the Select from Project interface, select the desired ligand structure, and click OK.
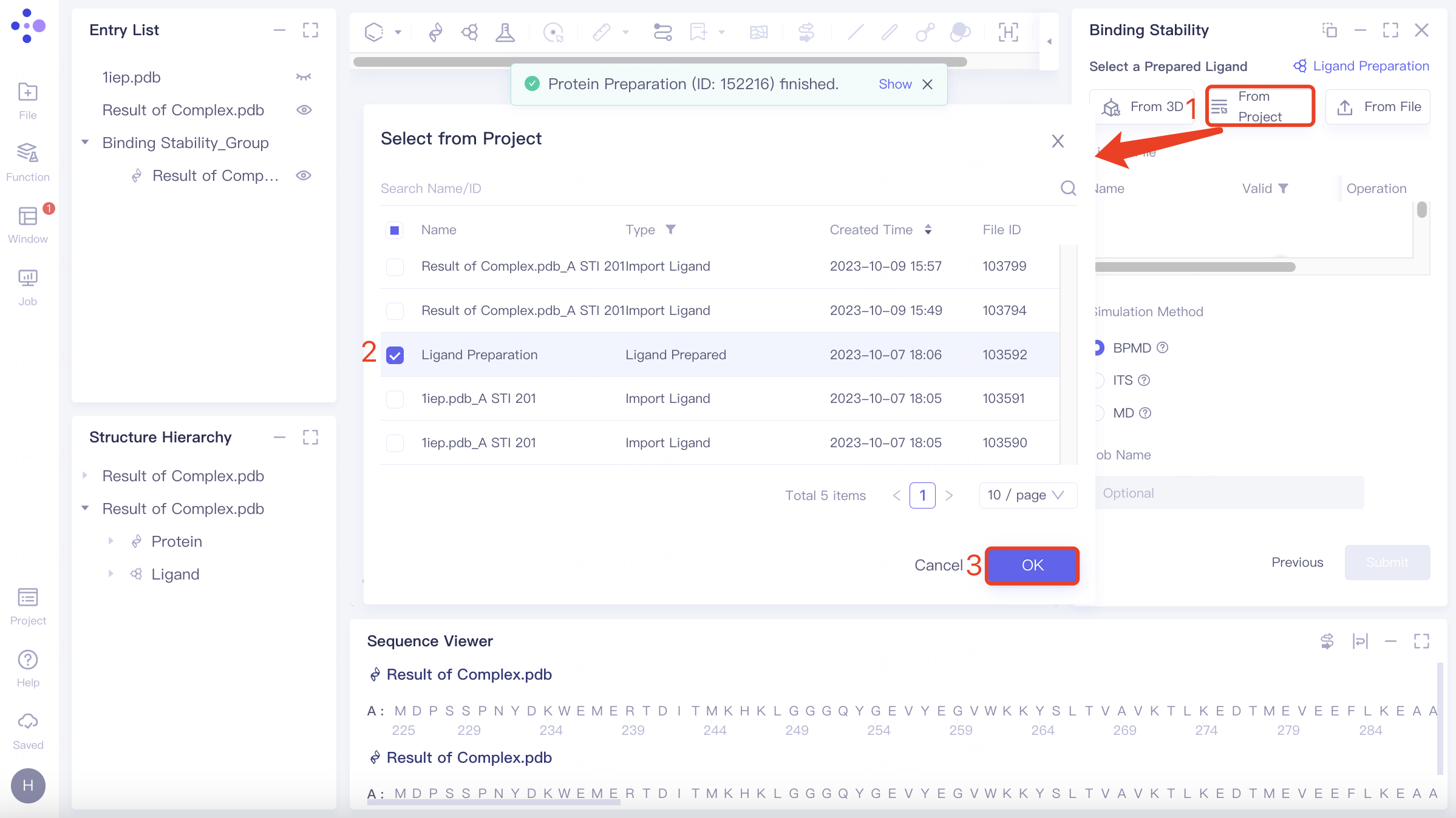
(3)Select File
Click the Select File checkbox → Select the desired ligand file (.mol and .sdf file formats supported) from the local folder and upload it.
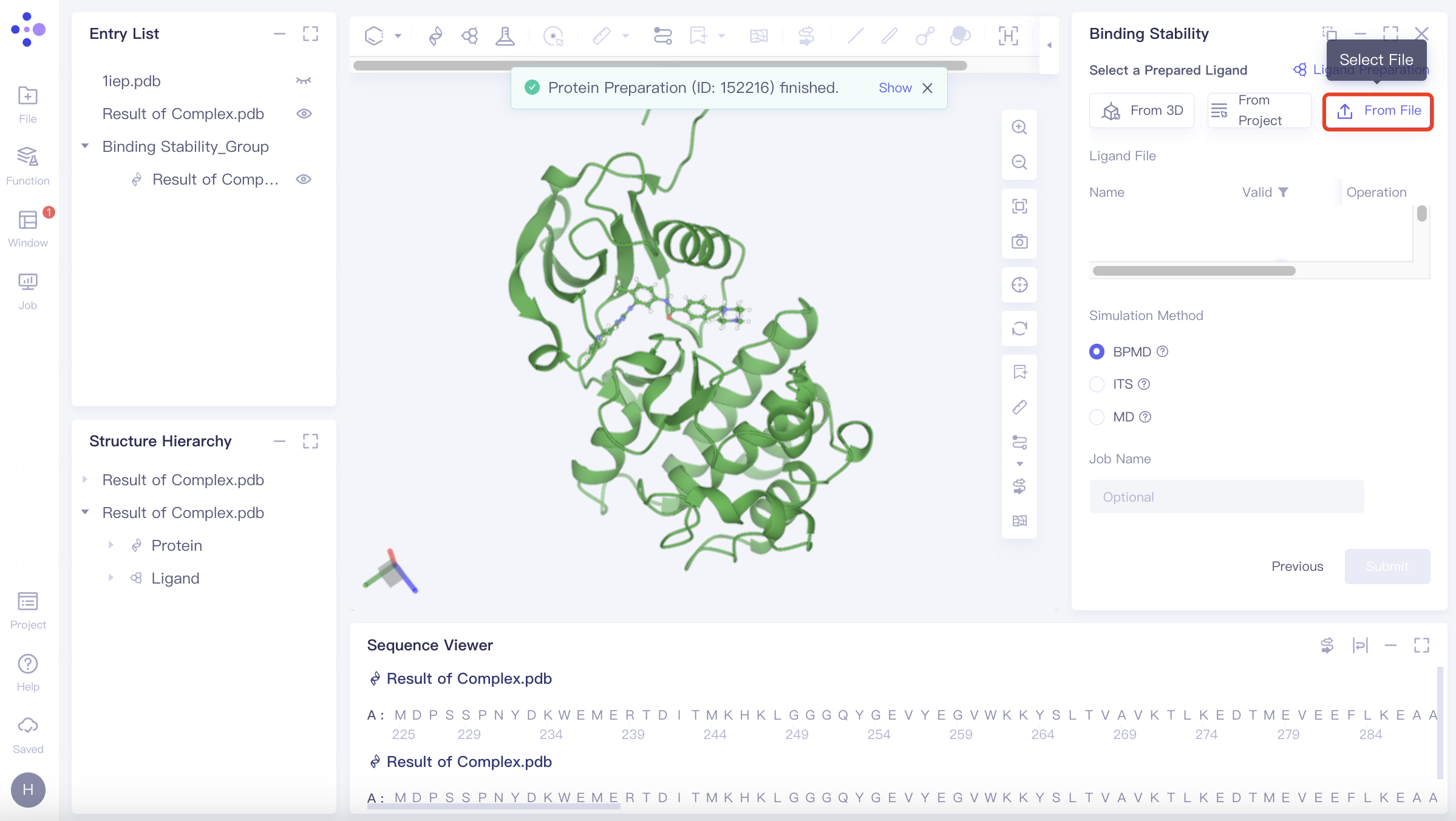
2.2.3 parameter setting and task submission
Simulation Method
BPMD: Binding pose meta dynamics, an improved sampling method that can effectively evaluate the stability of ligands in solution.
ITS: Integrated Tempering Sampling, a method for enhancing sampling over a wide range of energies and temperatures.
MD: Conventional Molecular Simulation, Traditional Molecular Dynamics.
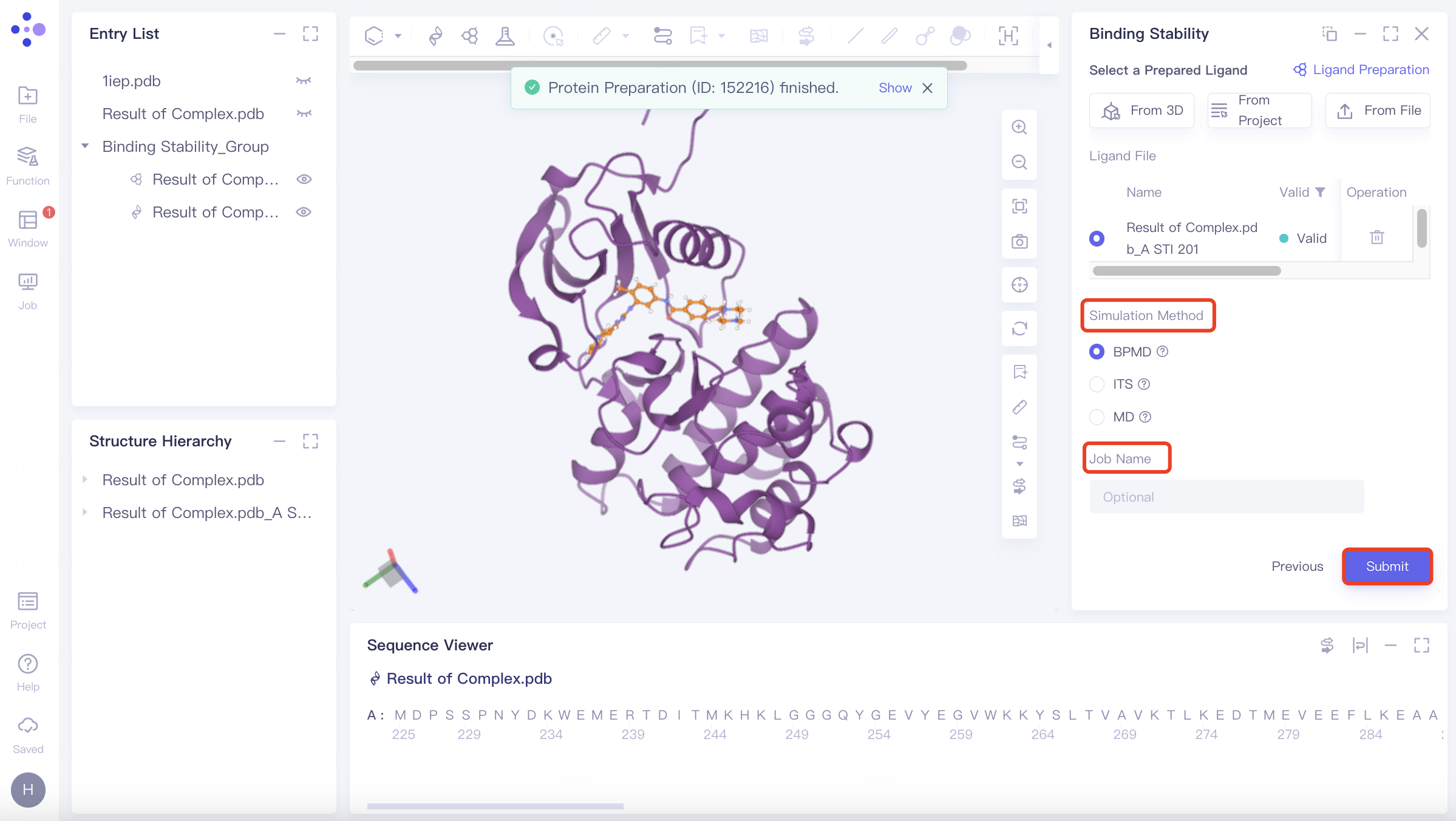
Name the task at "Job Name";
Click "Submit" to submit the task.
3. Results show
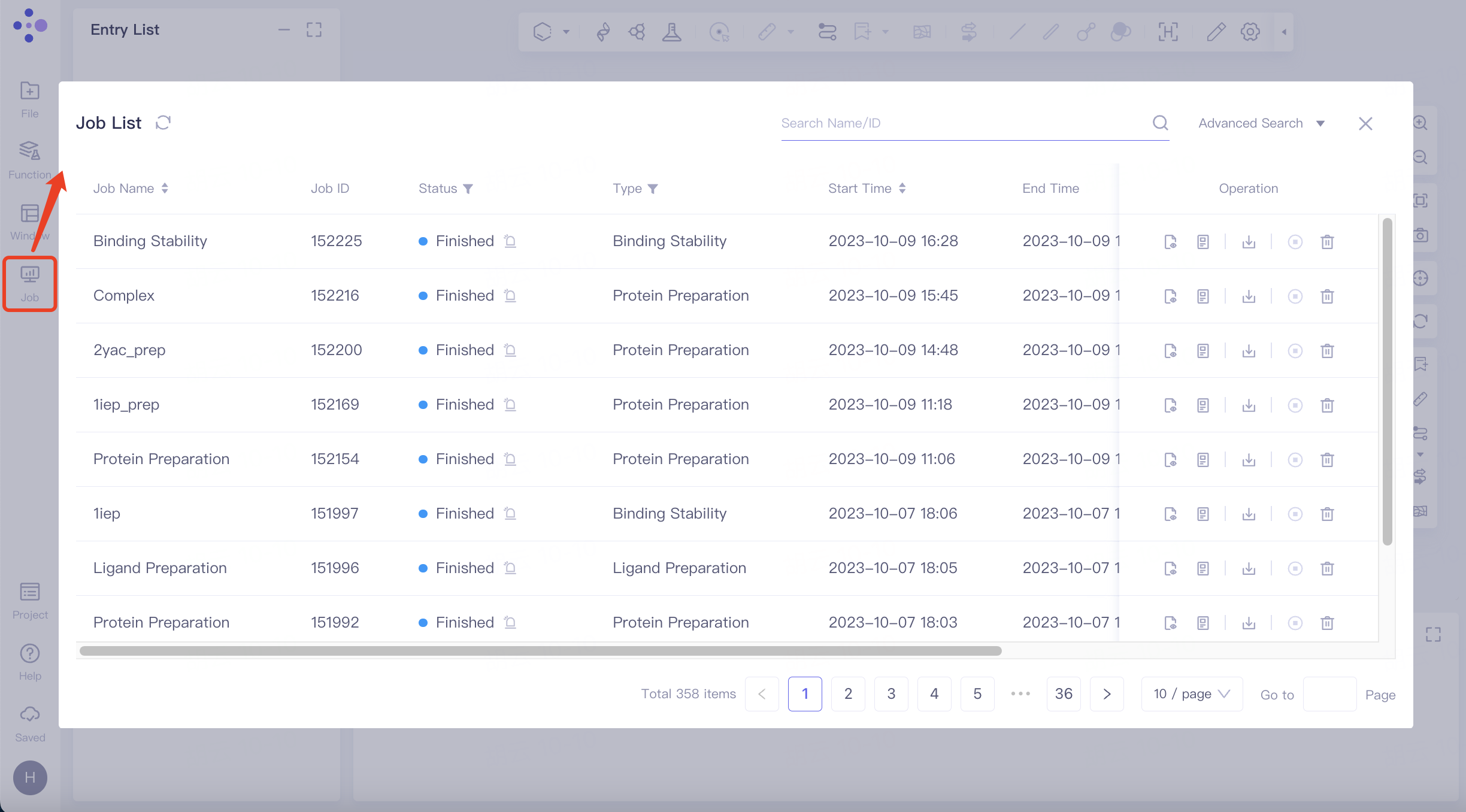
3.1 Entrance
The left general menu bar Job → Find the required task.
The task can be found by searching for Job Name or by filtering through Job Type.
3.2 Results
3.2.1 Results
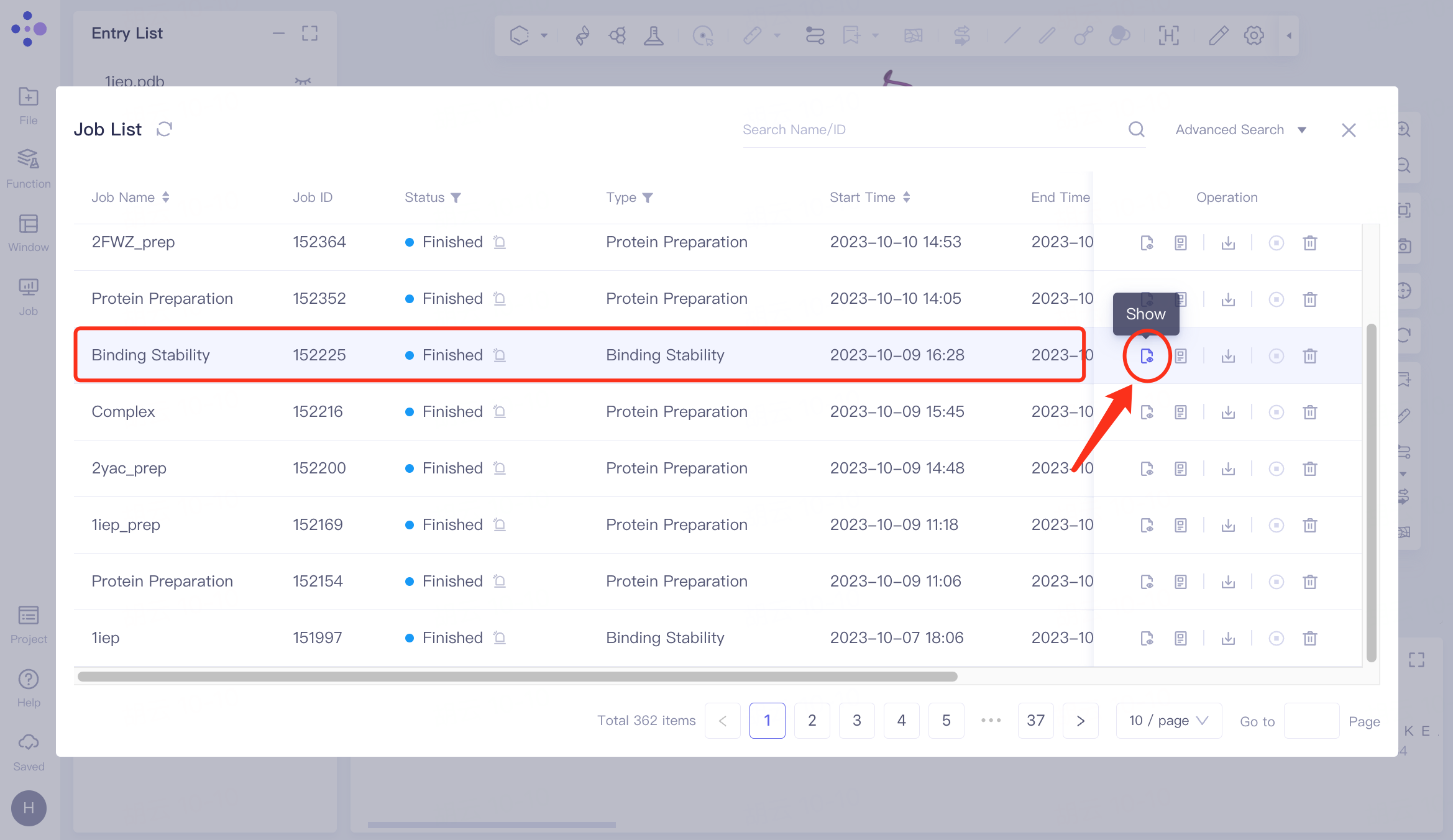
Select the task you want to view, and click Show in the Operation column to display the result of the task. The interface is shown in the left figure.
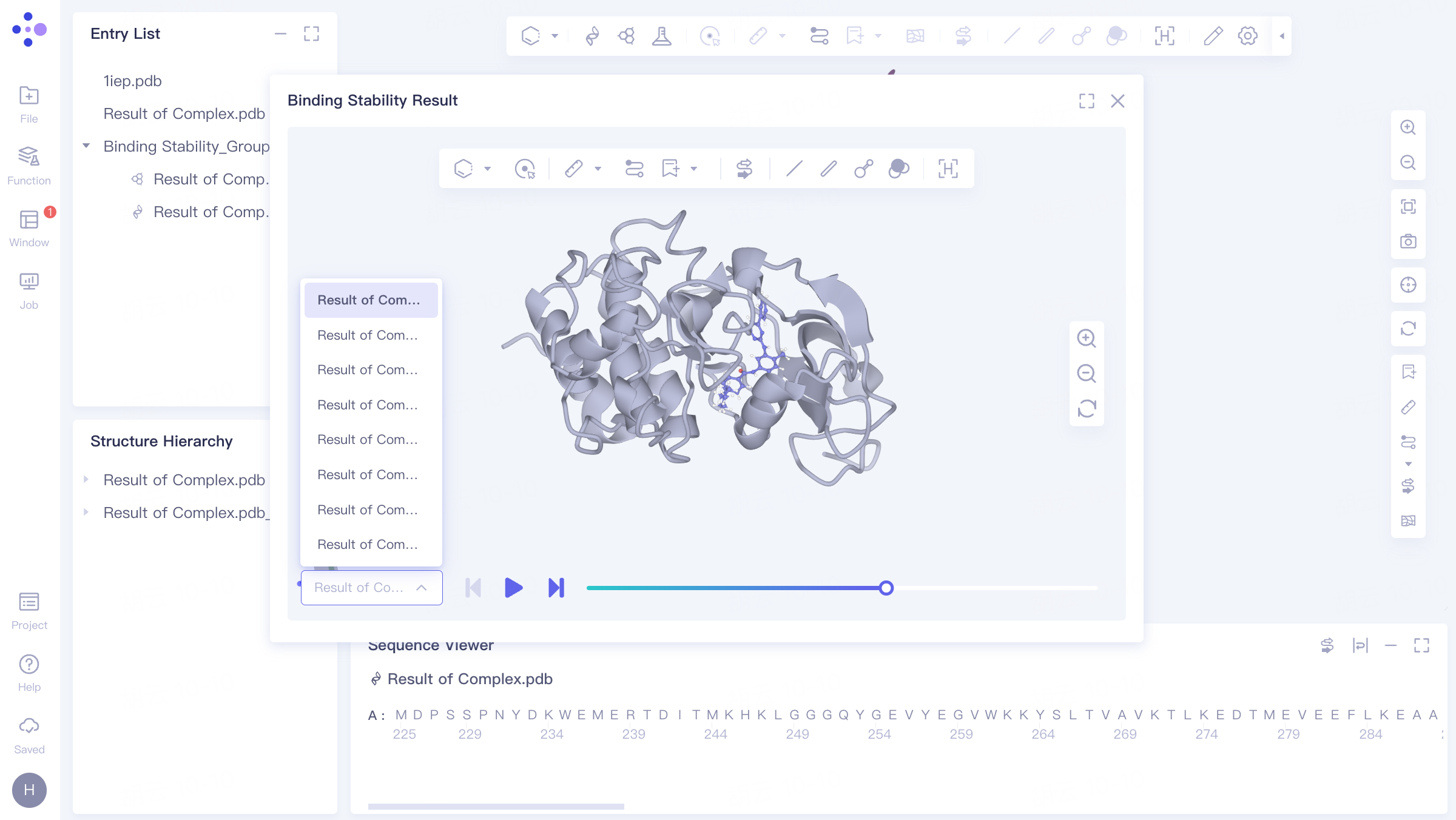
The dynamic trajectory of Binding Stability is shown in the video on the right
 |  |
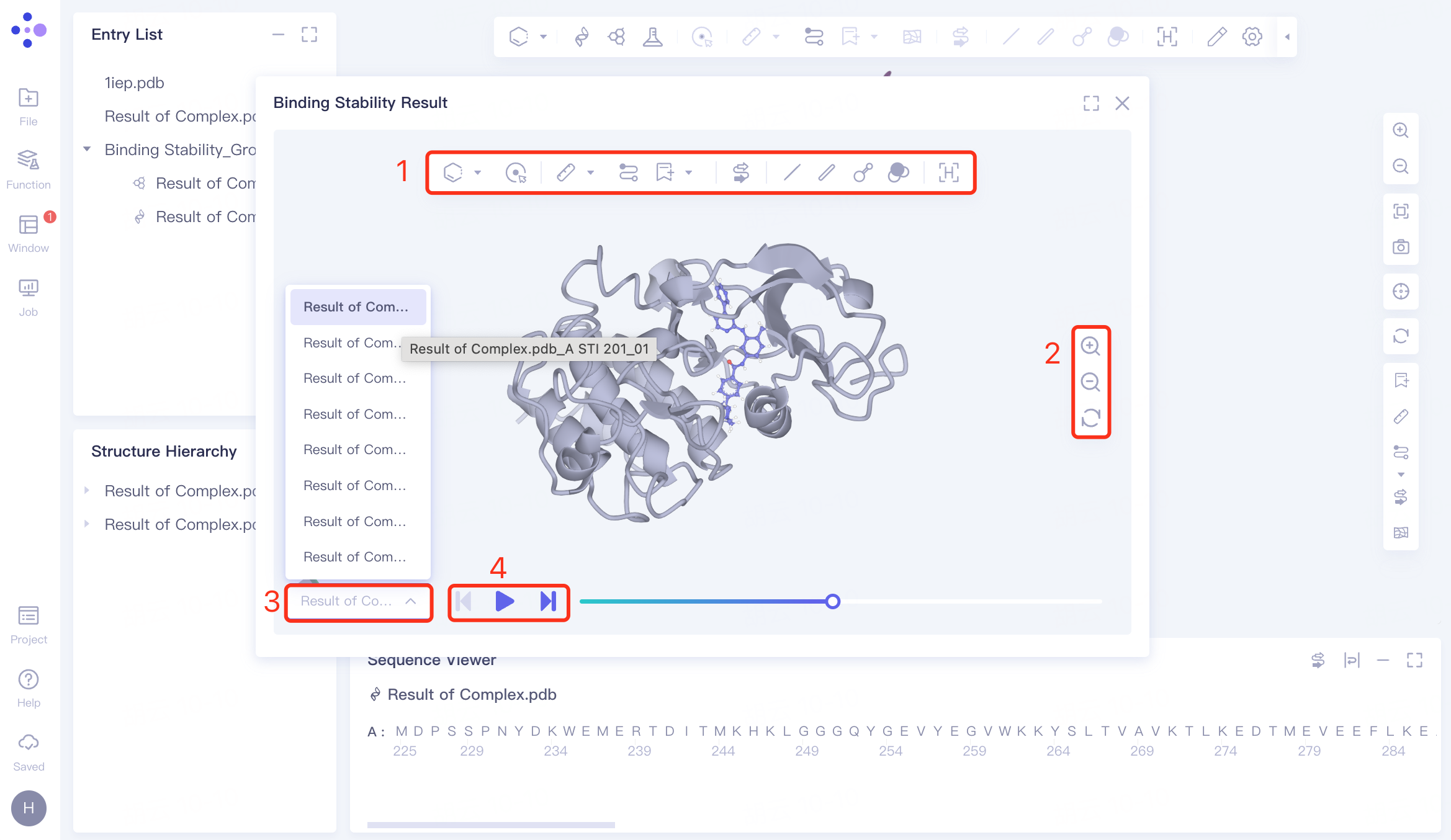
The video interface has four operation options:
1)View operation toolbar: refer to "Graphical Functions";
2)View universal tool: supports zooming in, zooming out, and refreshing the interface.
3) Track file list: It contains a total of 10 track files, which are 10 tracks with a duration of 10 ns for the input receptor and ligand .
4)Video operation tools: From left to right, they are "Switch to previous result", "Pause/Play", "Switch to next result".
3.2.2 download result description
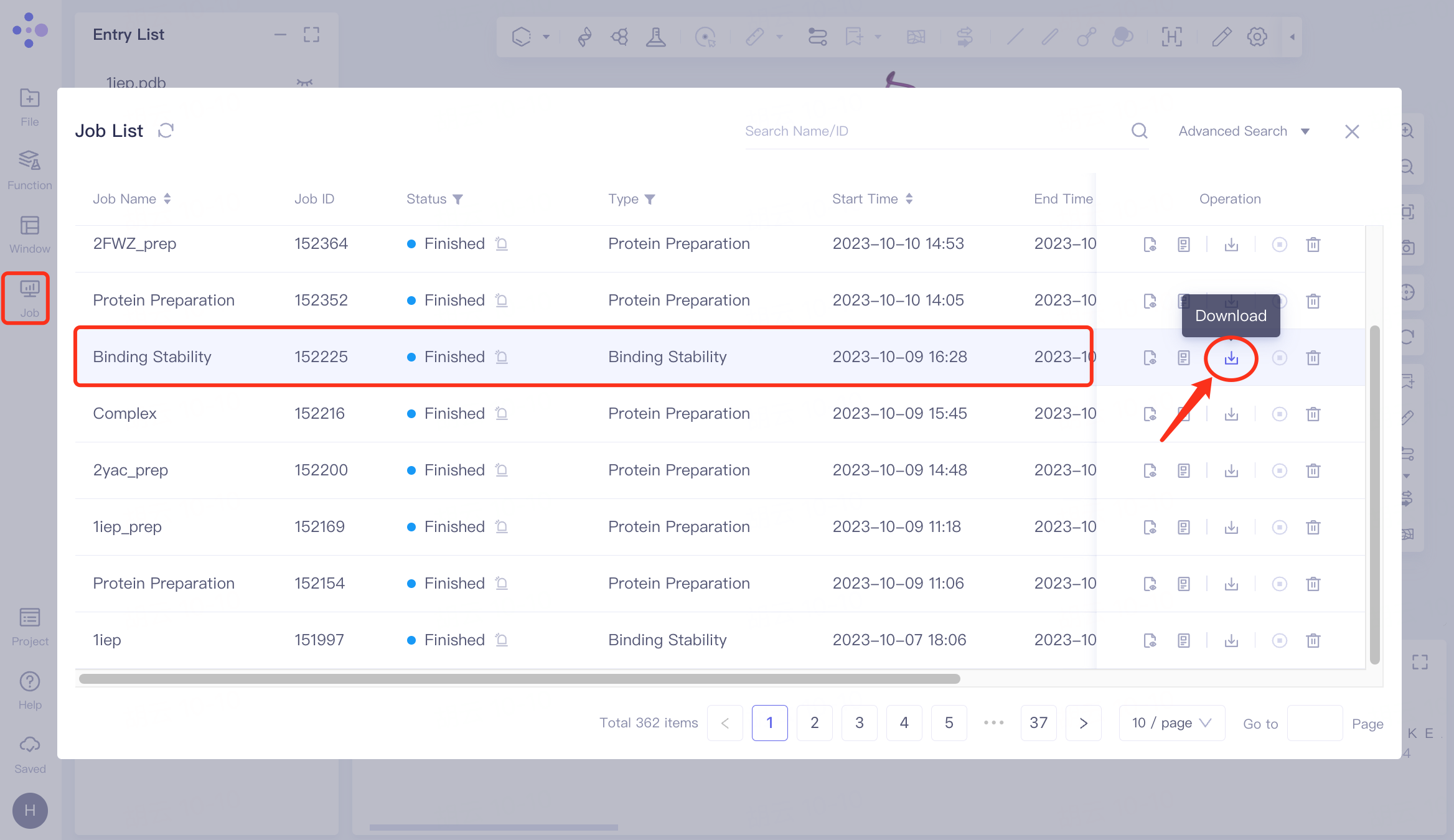
Download
Select the task to be downloaded, and click Download in the Operation column to download the result of the task. The interface is shown in the figure.
Download file instructions:
The resulting document provides the following information:
results.csv:
PoseScore: The average value of the root mean square deviation (RMSD) between the atom and the initial atom in 10 simulations.
ContactScore: The ratio of the number of interactions between the conformation per 100 ps and the previous 1 ns conformation is recorded as the ContactScore 'at each time. The ContactScore value in this table is the average of the ContactScore' in the last 2 ns of 10 simulations.
CompScore:In a single simulation, CompScore '= PoseScore - 5 * ContactScore. The CompScore in this table is recorded as the average of CompScore' in 10 simulations.
PoseScoreSD: Standard Deviation of PoseScore for 10 simulations.
ContactScoreSD: Standard Deviation of ContactScore for 10 simulations.
CompScoreSD: Standard Deviation of CompScoreSD for 10 simulations.
rep_0~ rep_9: Tracks folder with two files
.Xtc file: Molecular dynamics trajectory file.
.Pdb file: Receptor- ligand complex file.
.sdf, .pdb files outside the track folder: input small molecule and protein files.
.png file: Changes of PoseScore, ContactScore, and CompScore over a period of 10 ns.
GBSA.csv file: GBSA free energy values under different frames.
.txt file: log file.