Molecular Docking Based on Hydrogen Bond Interaction
Introduction
1A4K is the three-dimensional structure of an antibody (39-A11) that catalyzes the Diels-Alder reaction. The antibody catalyzes the reaction through a combination of packaging and hydrogen bonding interactions that control the relative geometry of the binding substrate and the electron distribution in the diphilic reagent [1].
This tutorial is based on the Docking module of the Hermite platform to re-dock 1A4K eutectic based on hydrogen bond restriction - .
1. Introduction of eutectic structure and extraction of ligands
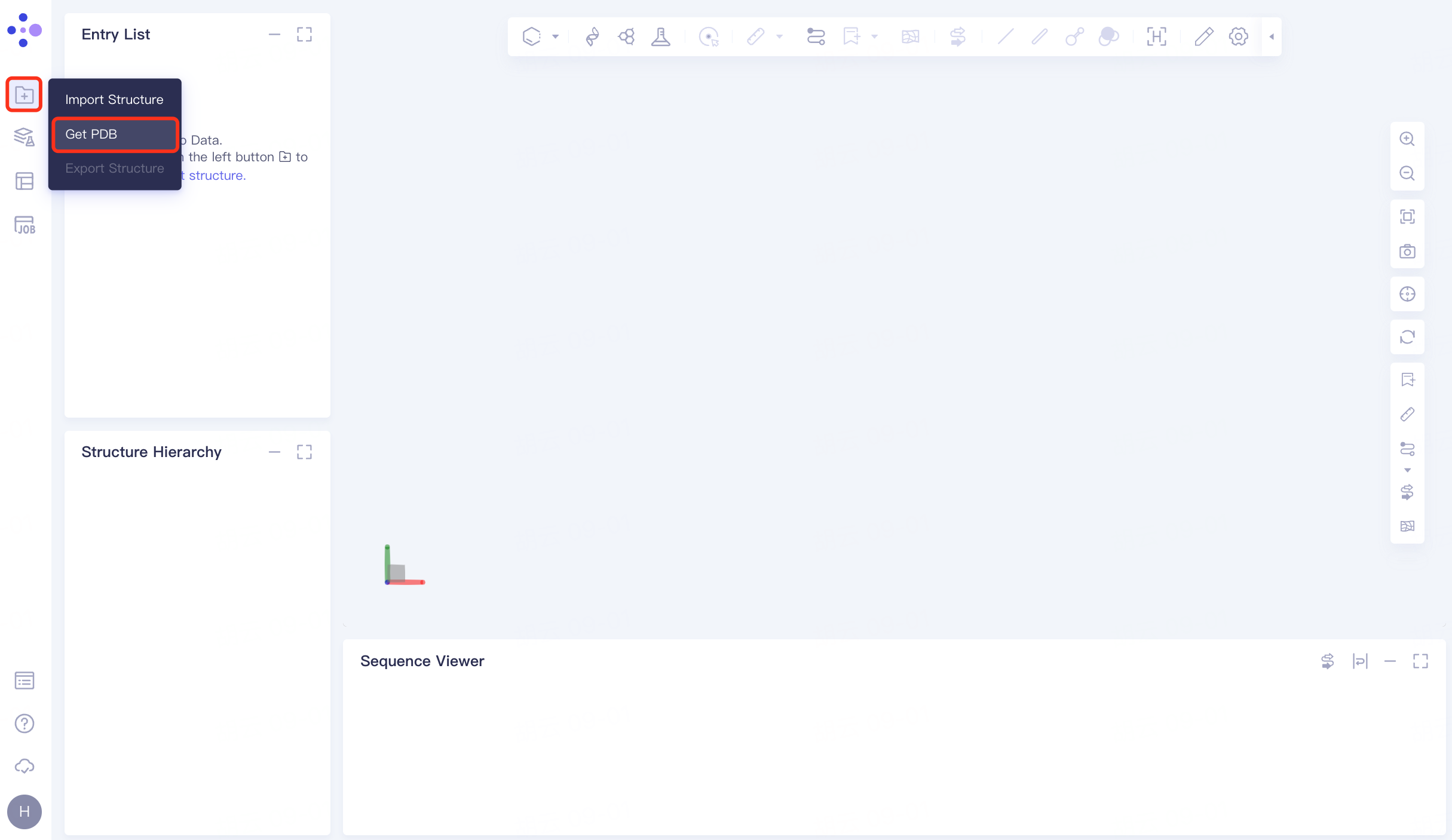
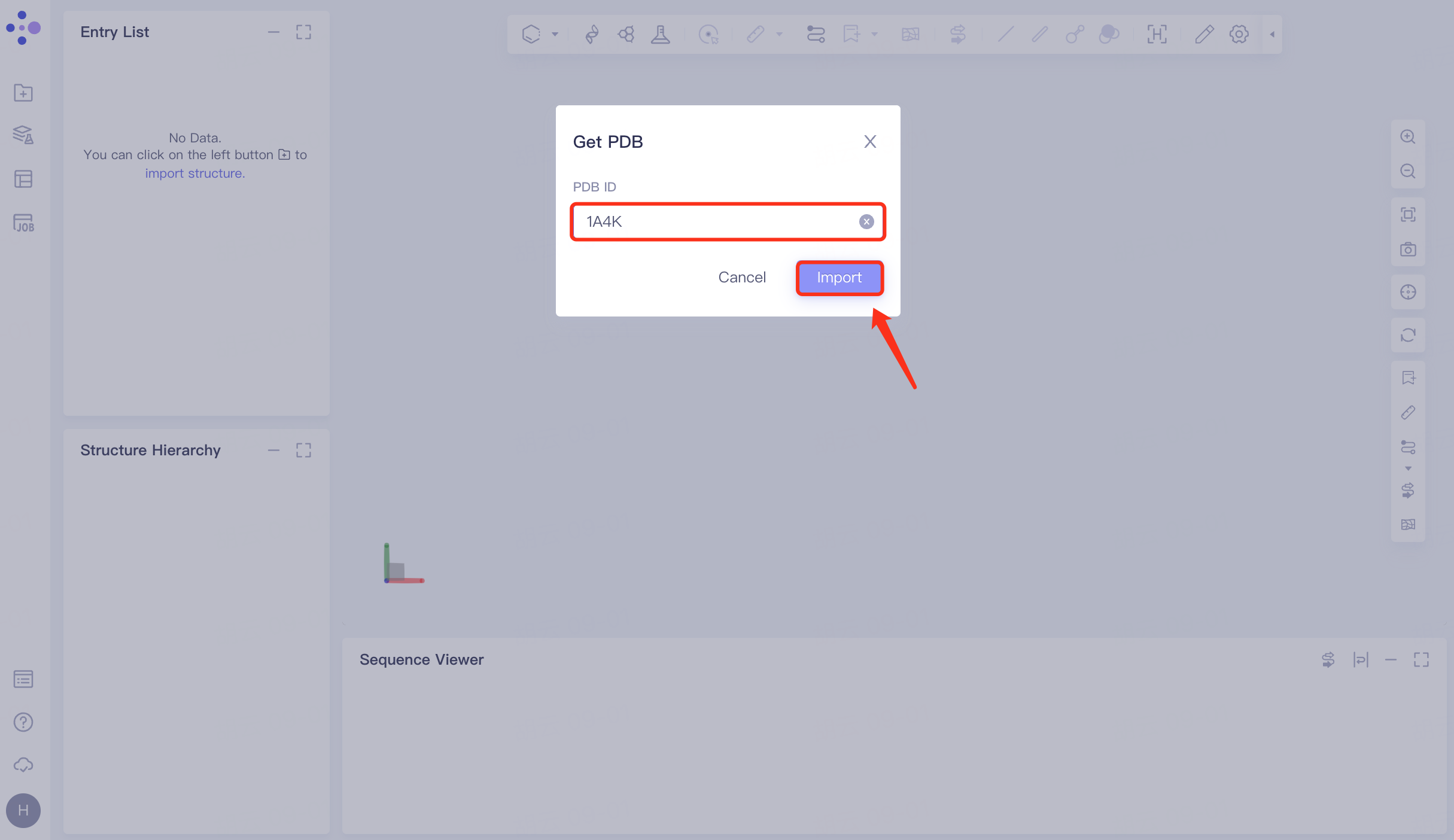
1.1 Get PDB
In the left general menu bar File → Get PDB → pop-up window, enter the PDB ID: 1A4K → Import.
 |  |
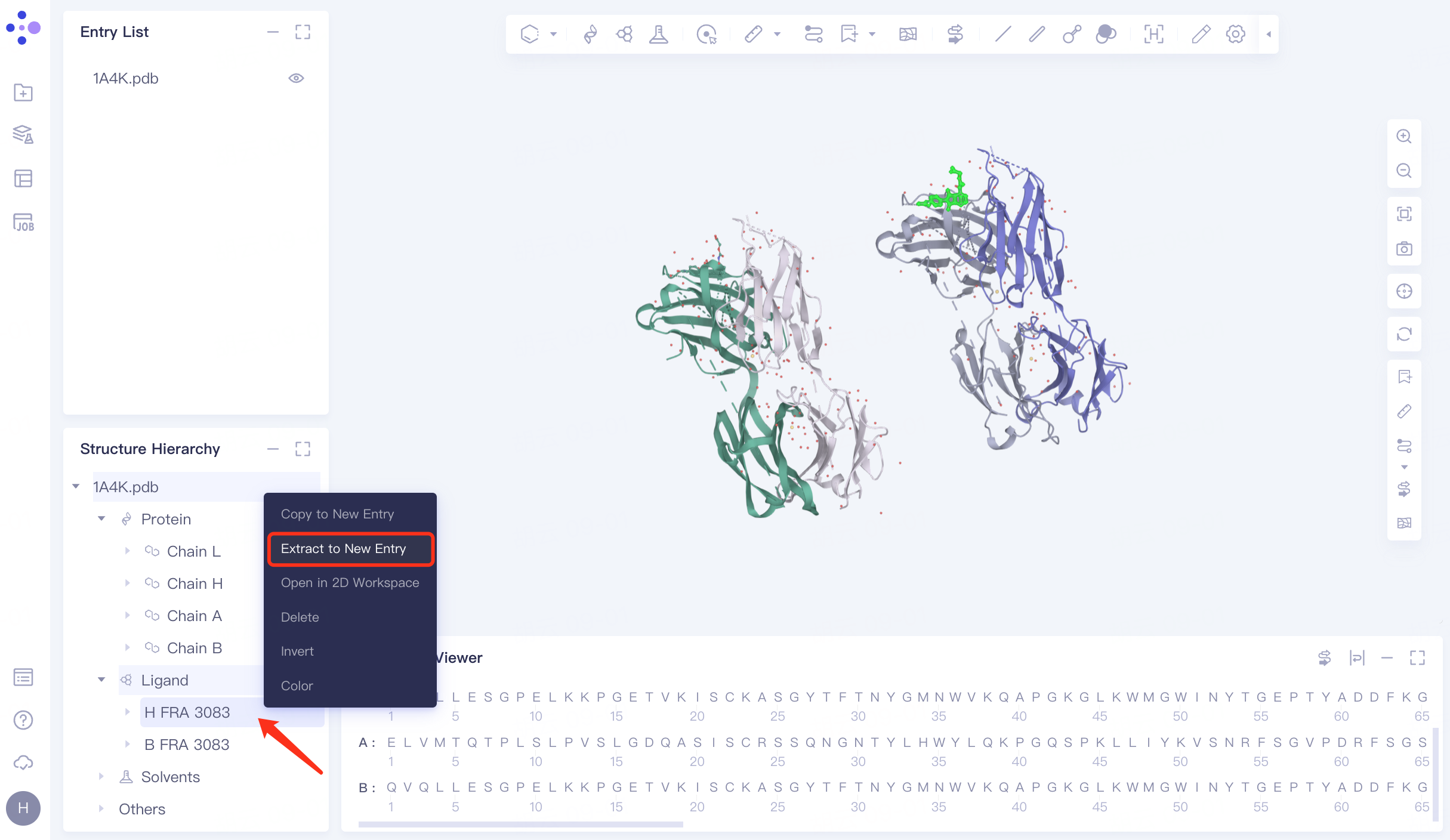
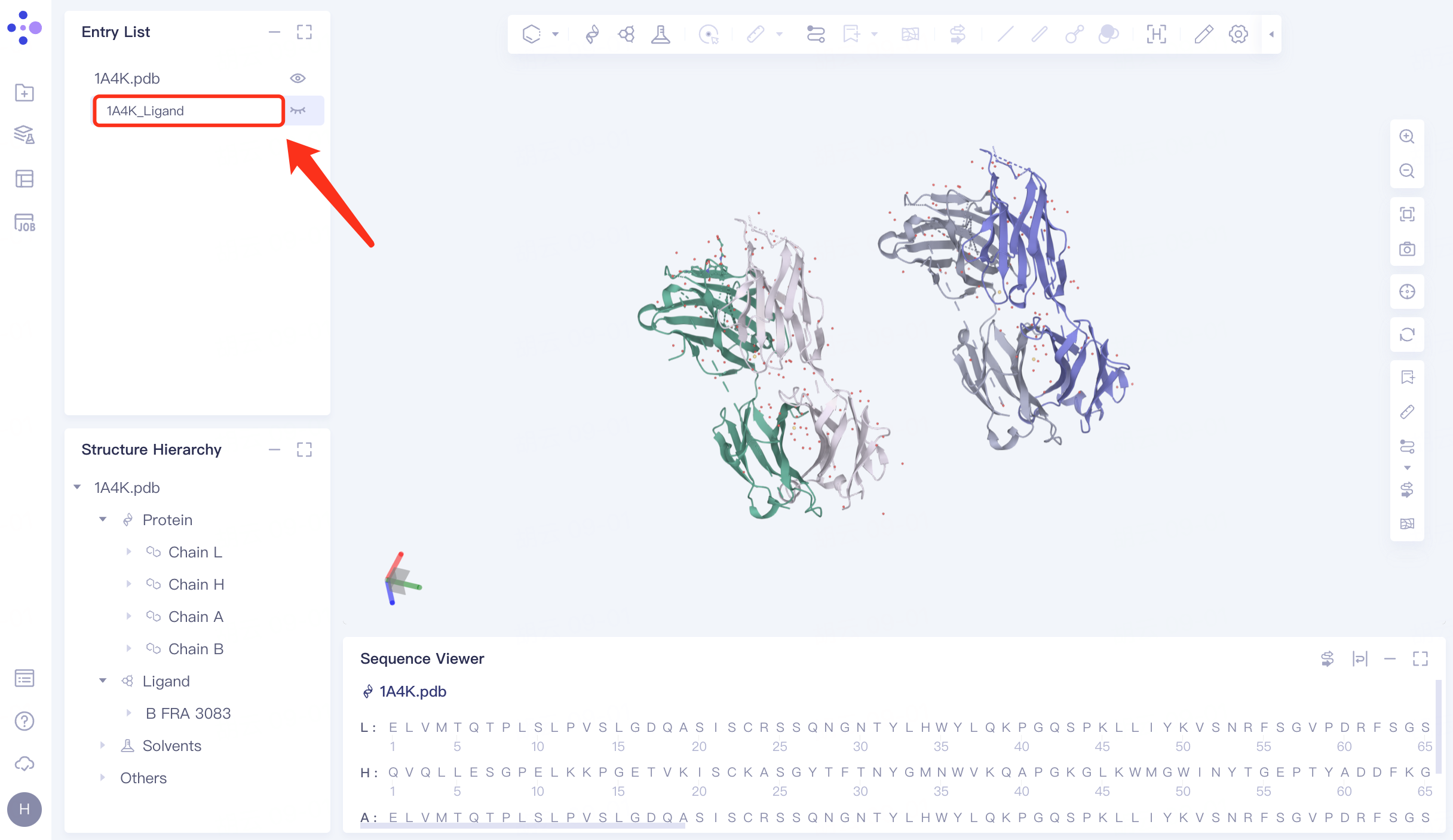
1.2 Extraction of ligand molecules
In the Structure Hierarchy window, expand the structure of 1A4K.pdb → select the ligand molecule "HFRA 3083", right click → Extract to New Entry → The extracted ligand molecule appears in the Entry List window and is renamed to "1A4K_Ligand".
1A4K contains two antibody complexes .
 |  |
1.3 Protein file processing
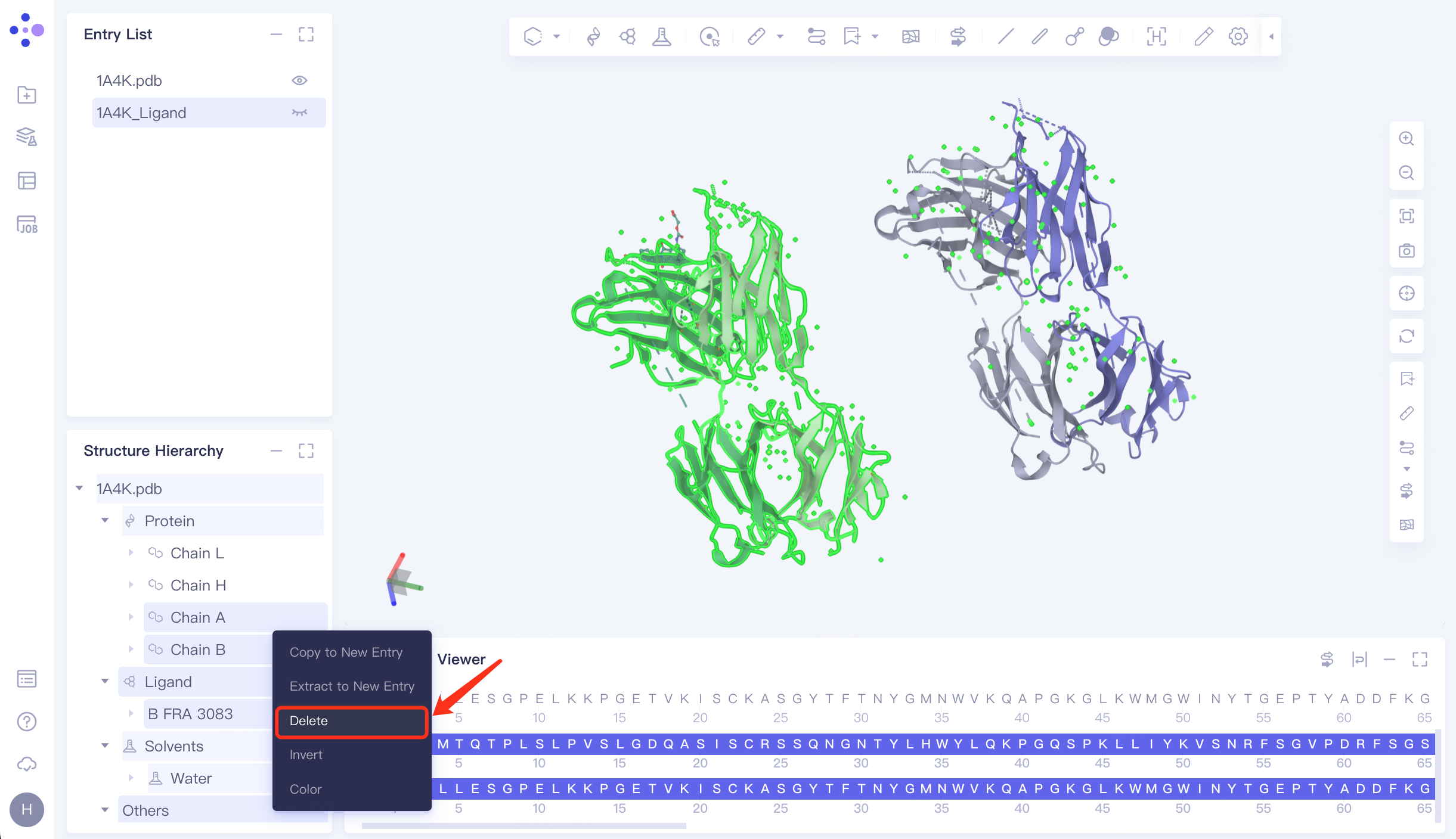
Select the proteins Chain A, Chain B, Ligand , Solvents and Others in the Structure Hierarchy window, right click and select "Delete".
The processed structure is shown in FIG.
 | 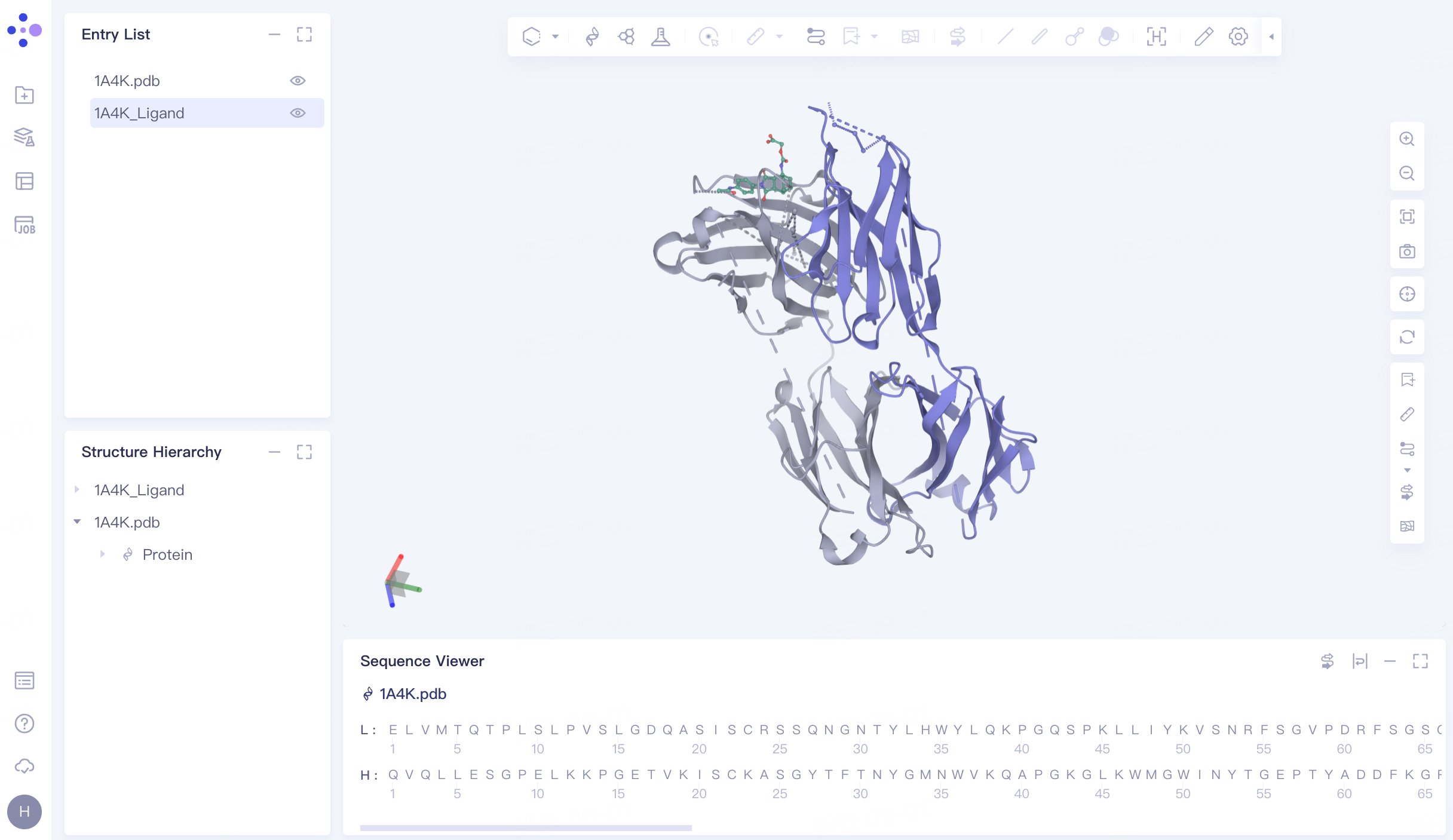 |
2. Protein Preparation
2.1 Protein Preparation
The left general menu bar Function → Structure Modeling → Protein Preparation. The right interface appears.
 |  |
2.2 Selection of protein structures
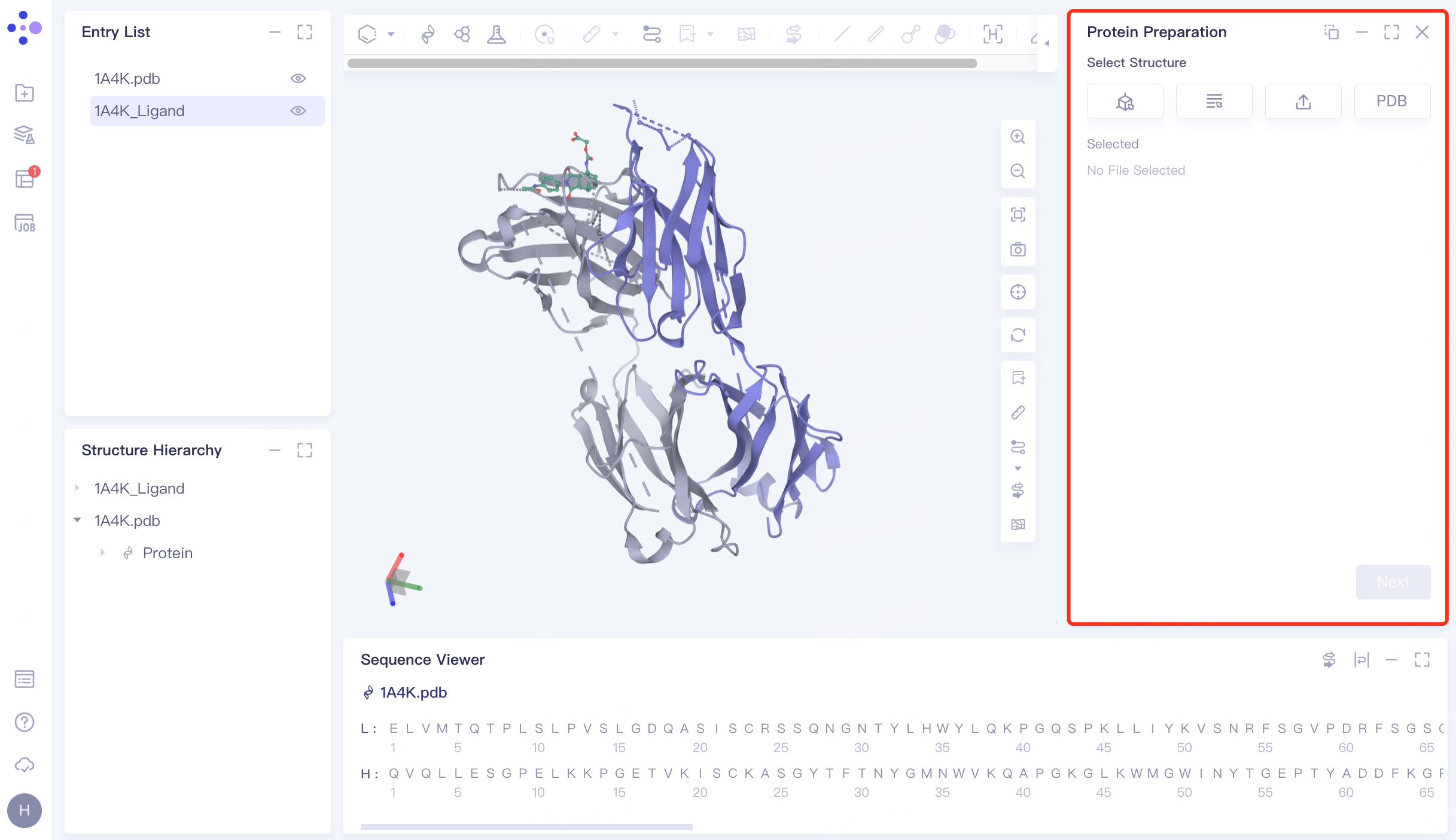
Click the Select from 3D Workspace checkbox → pop-up Select Structure interface → Select 1A4K Protein in the Structure Hierarchy window on the left side of the interface → Select Structure box to display the selected protein name, click OK.
Click "Next" to go to the next step.
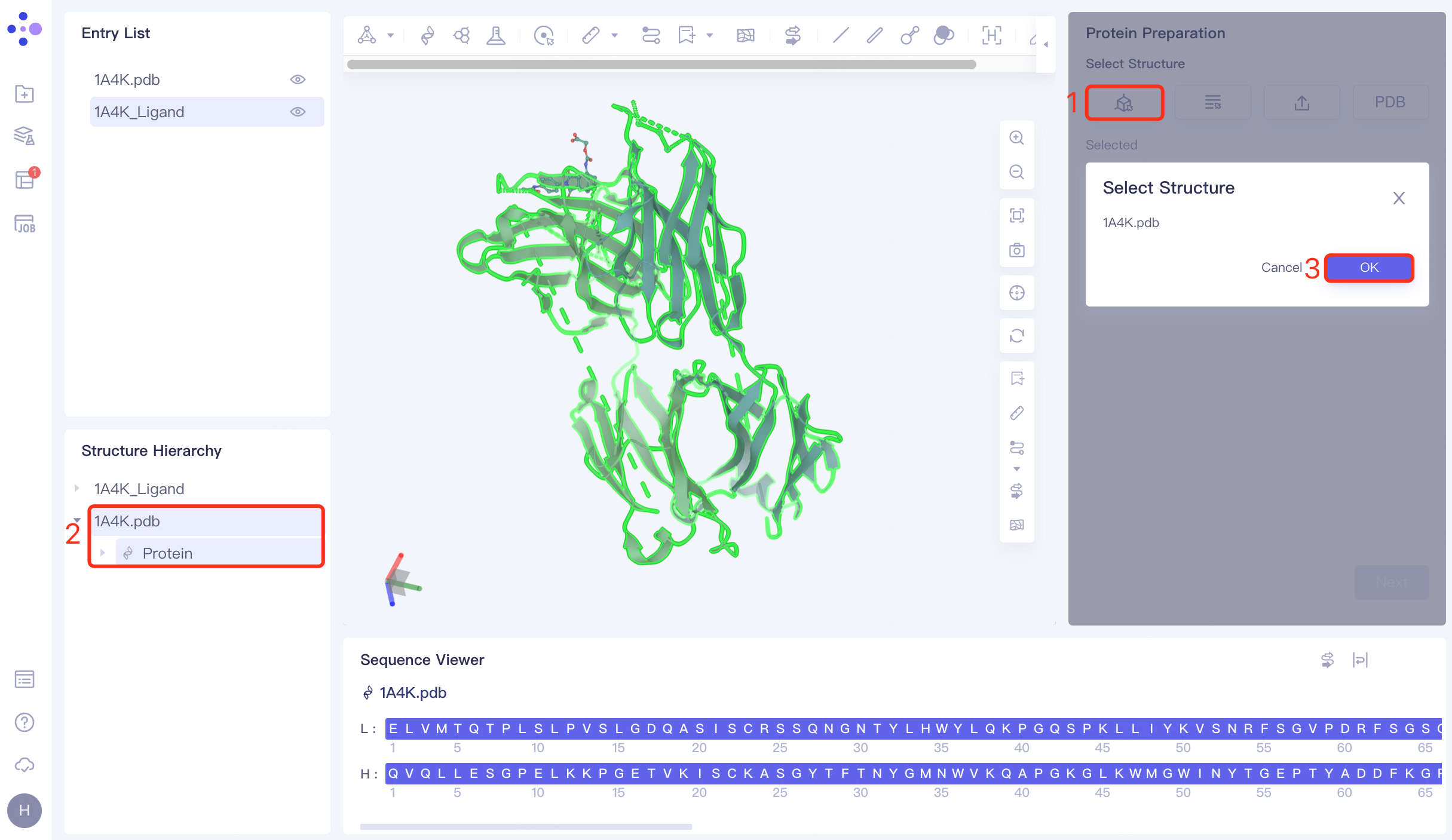
2.3 Select the Polymer to Keep
Keep Chain L and Chain H.
Click "Next" to go to the next step.
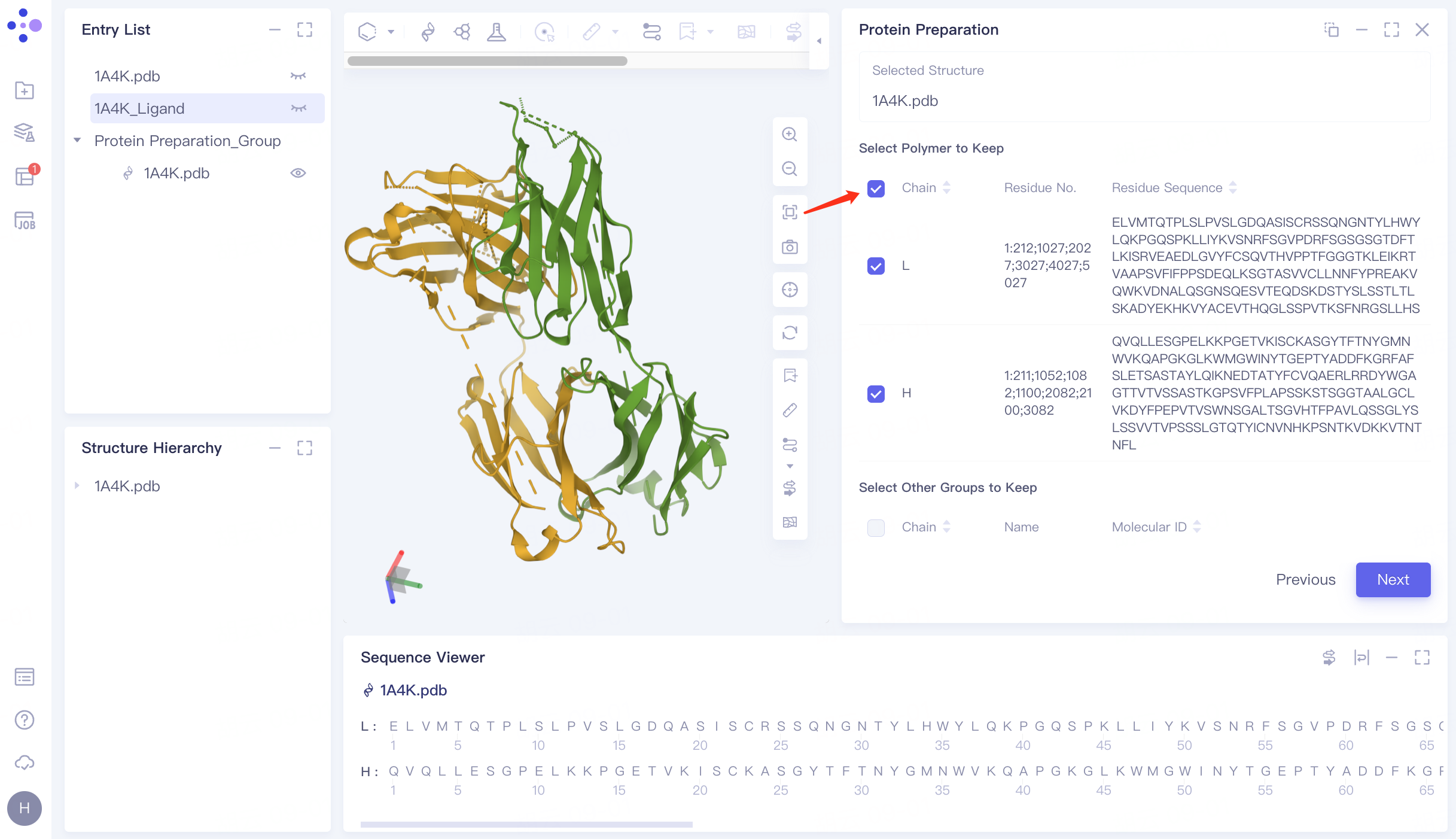
2.4 Parameter settings
Select Missing Residues to Repair: Not selected here (the missing amino acid is not in the critical region) .
Prepared Settings
Add Missing Side Chains: Check to add missing side chains;
Add Hydrogens: Check, Hydrogenation;
Modify Protonation State: Check, adjust the protein environment pH to physiological pH 7.4;
Optimize Hydrogen Bonding Network: Check to optimize Hydrogen Bonding Network;
Energy Minimization: Uncheck, no energy minimization is performed here.
Name the task "1A4K_Protein_pre" at Job Name.
Click "Submit" to submit the task.
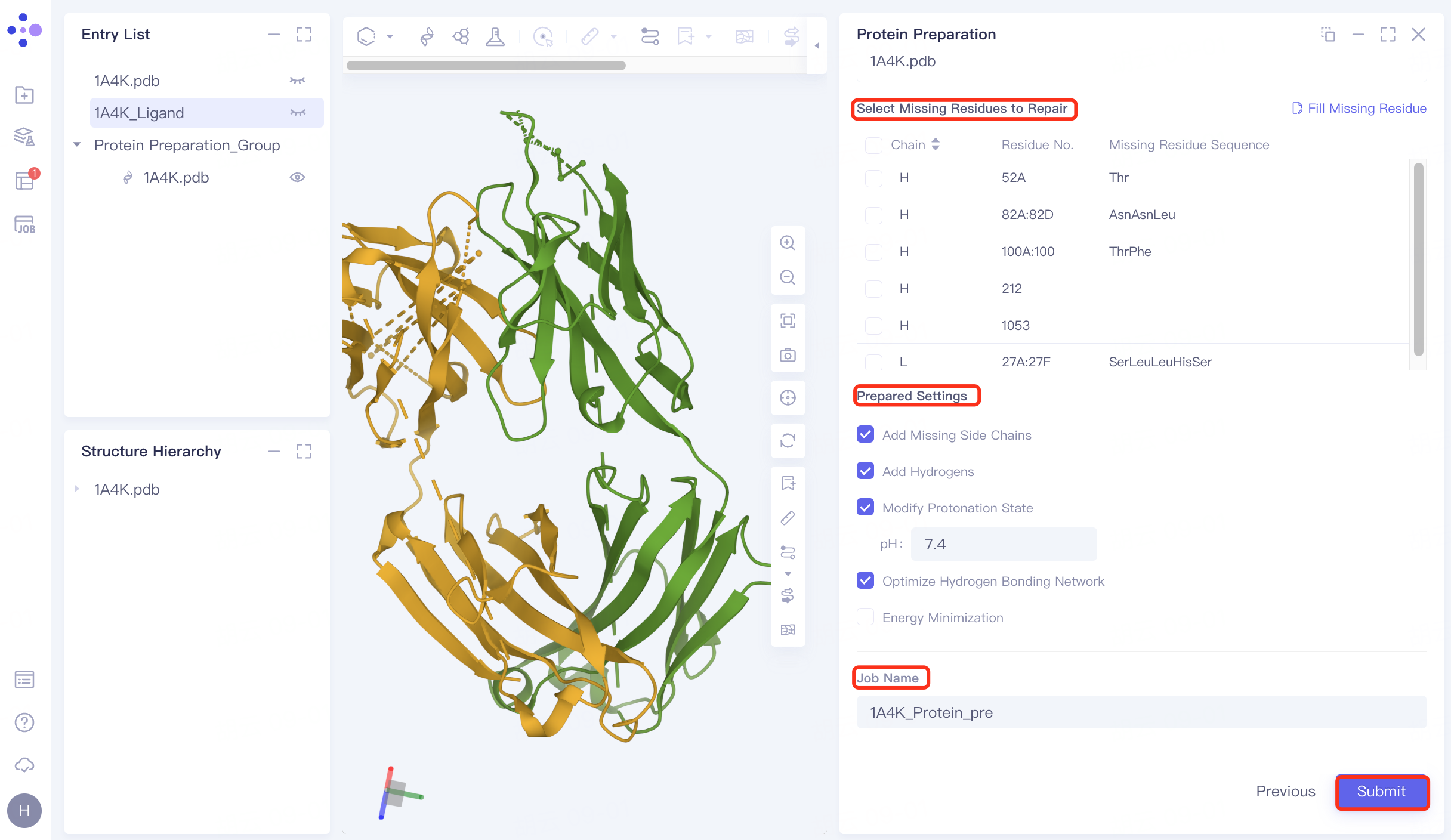
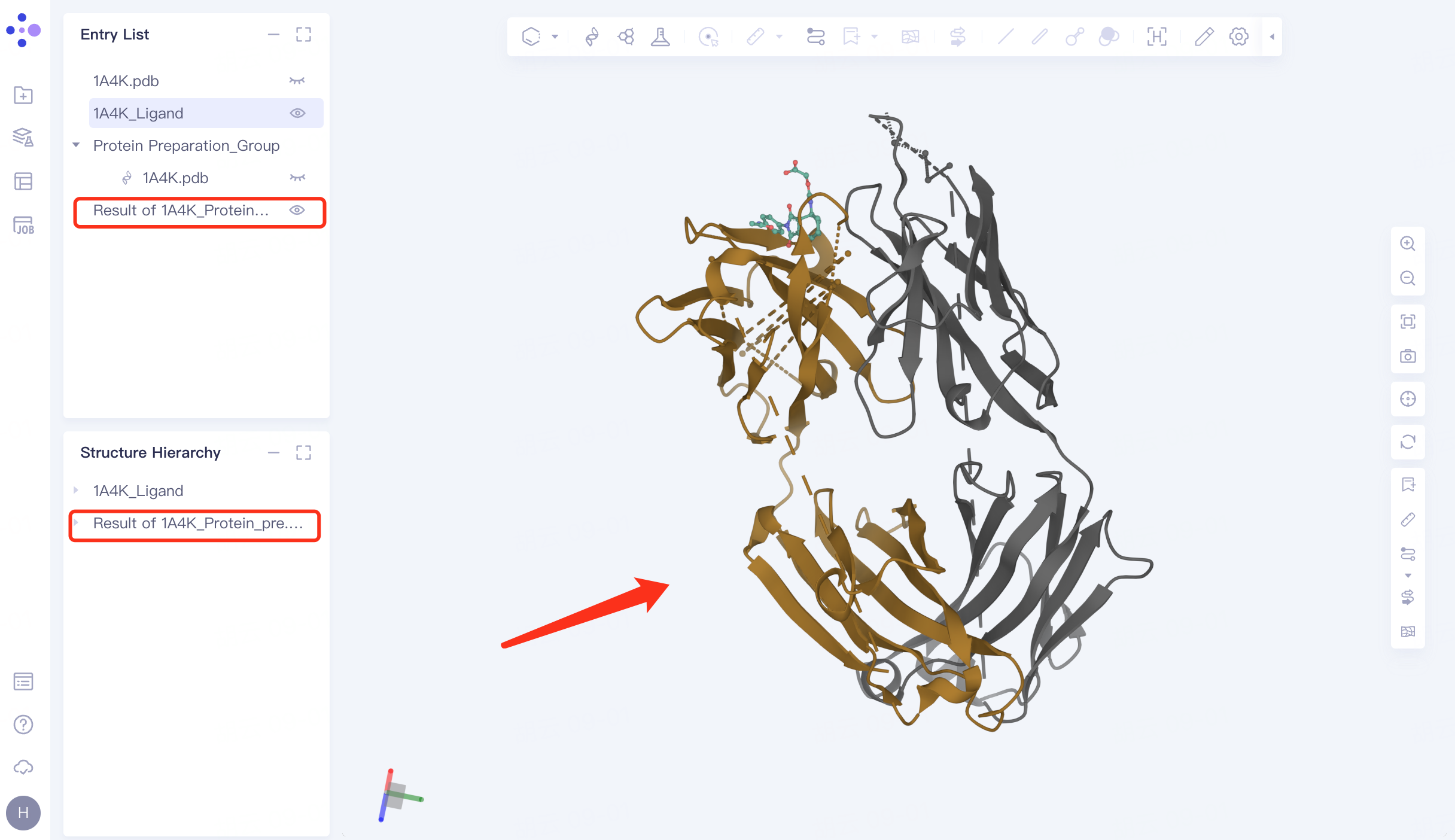
2.5 Checking for protein
Click Job in the left general menu bar → find the "1A4K_Protein_pre" task in the pop-up Job List interface → click Show.
The prepared protein structure is shown in the figure.
 |  |
3. Ligand Preparation
3.1 Entrance
The left general menu bar Function → Virtual Screening → Ligand Preparation. The right interface appears.
 | 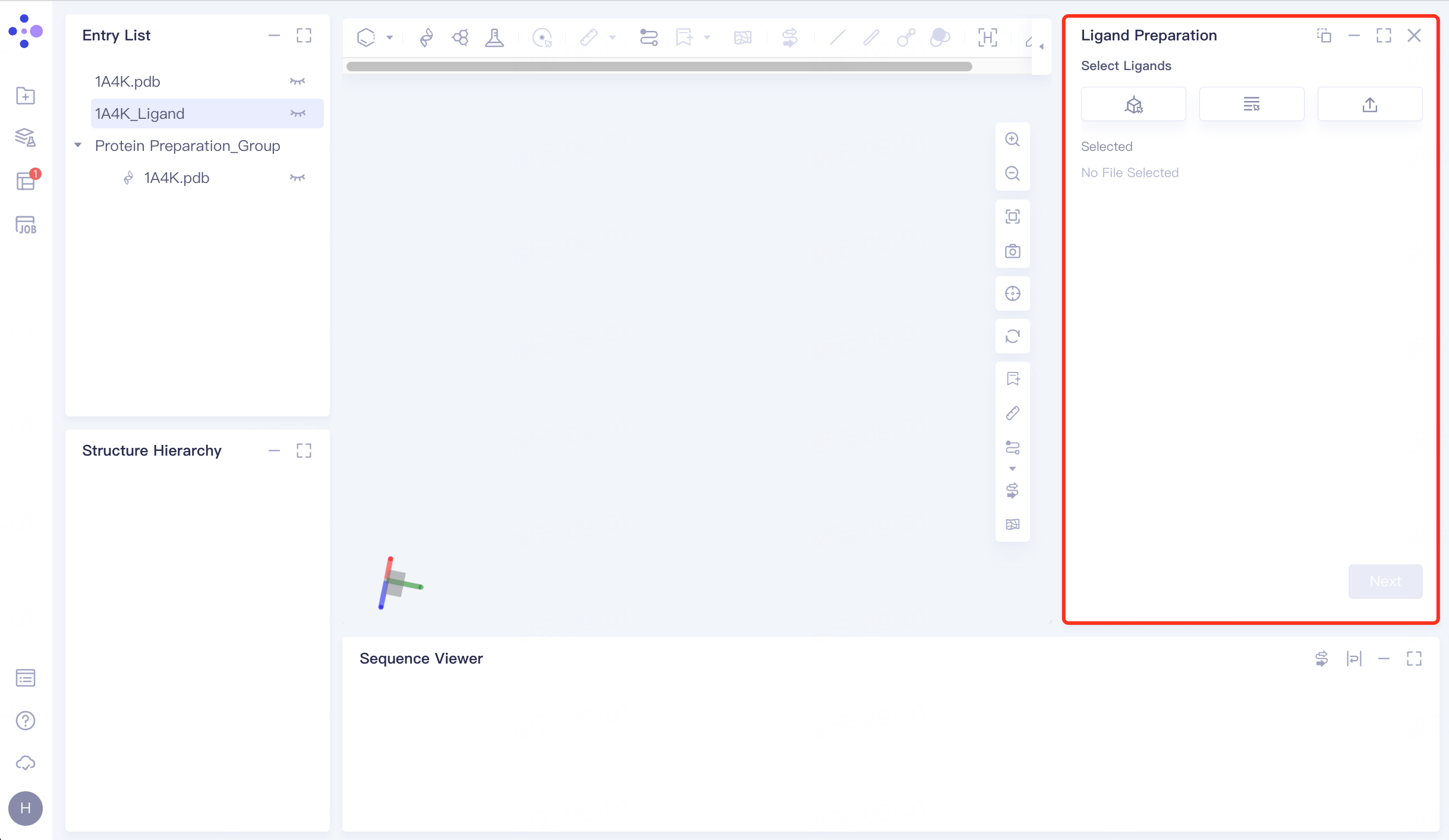 |
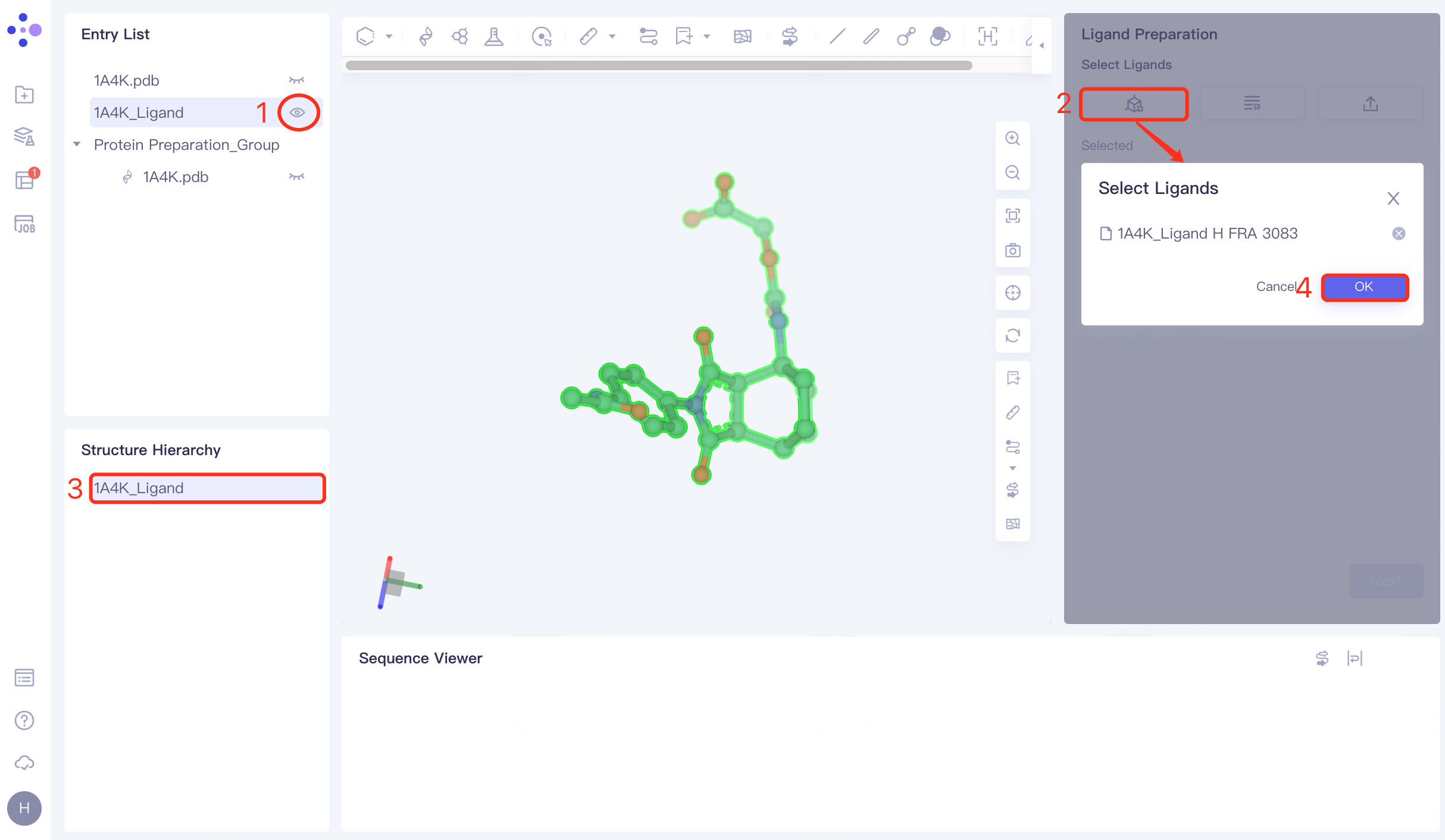
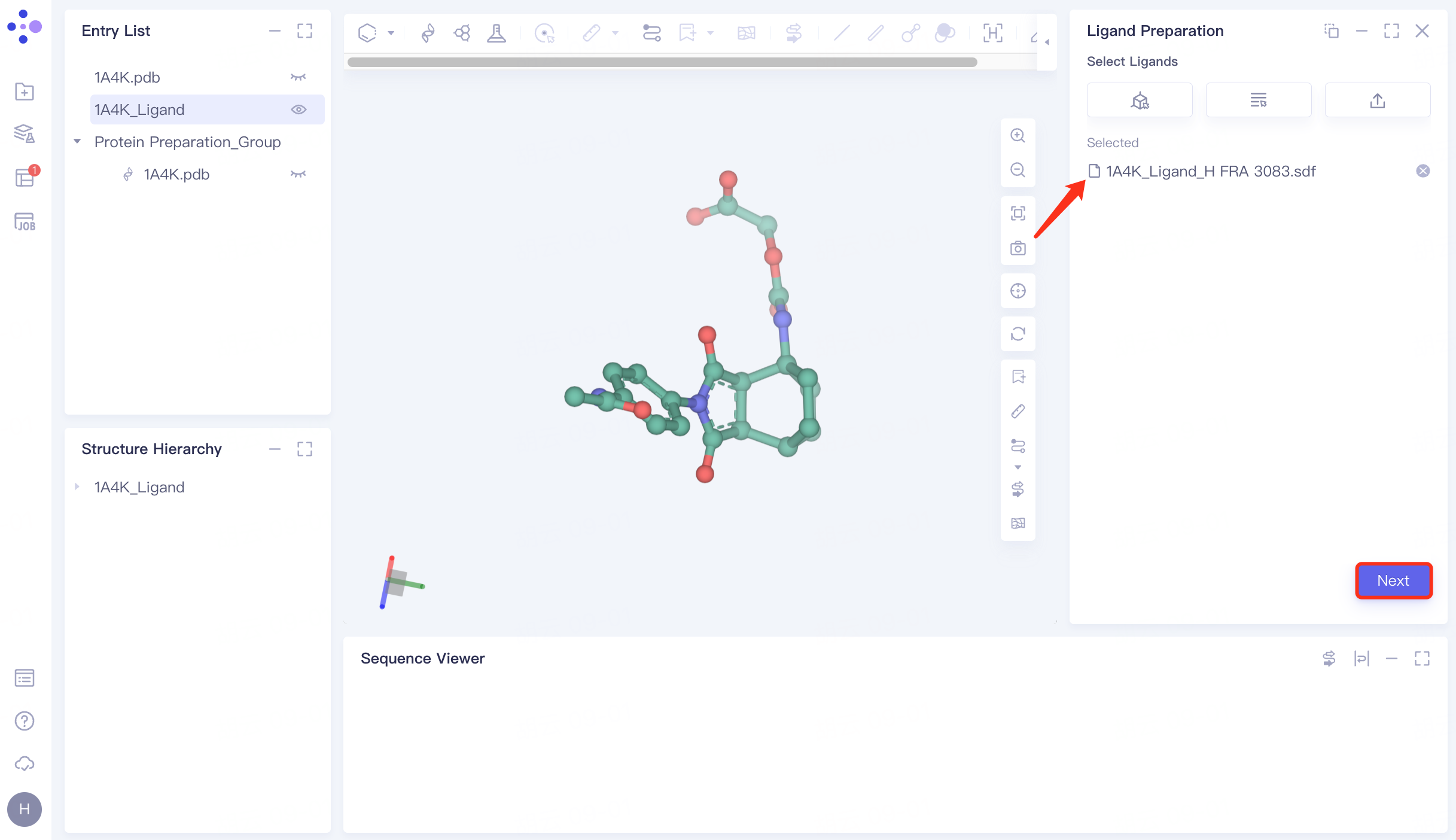
3.2 Selection of ligand structures
The Entry List window adjusts the 1A4K_Ligand to the display state → Click the From 3D Workspace checkbox → The Select Ligands window pops up → The Structure Hierarchy window selects the ligand molecule → The Select Ligands window displays the selected ligand name, and click OK.
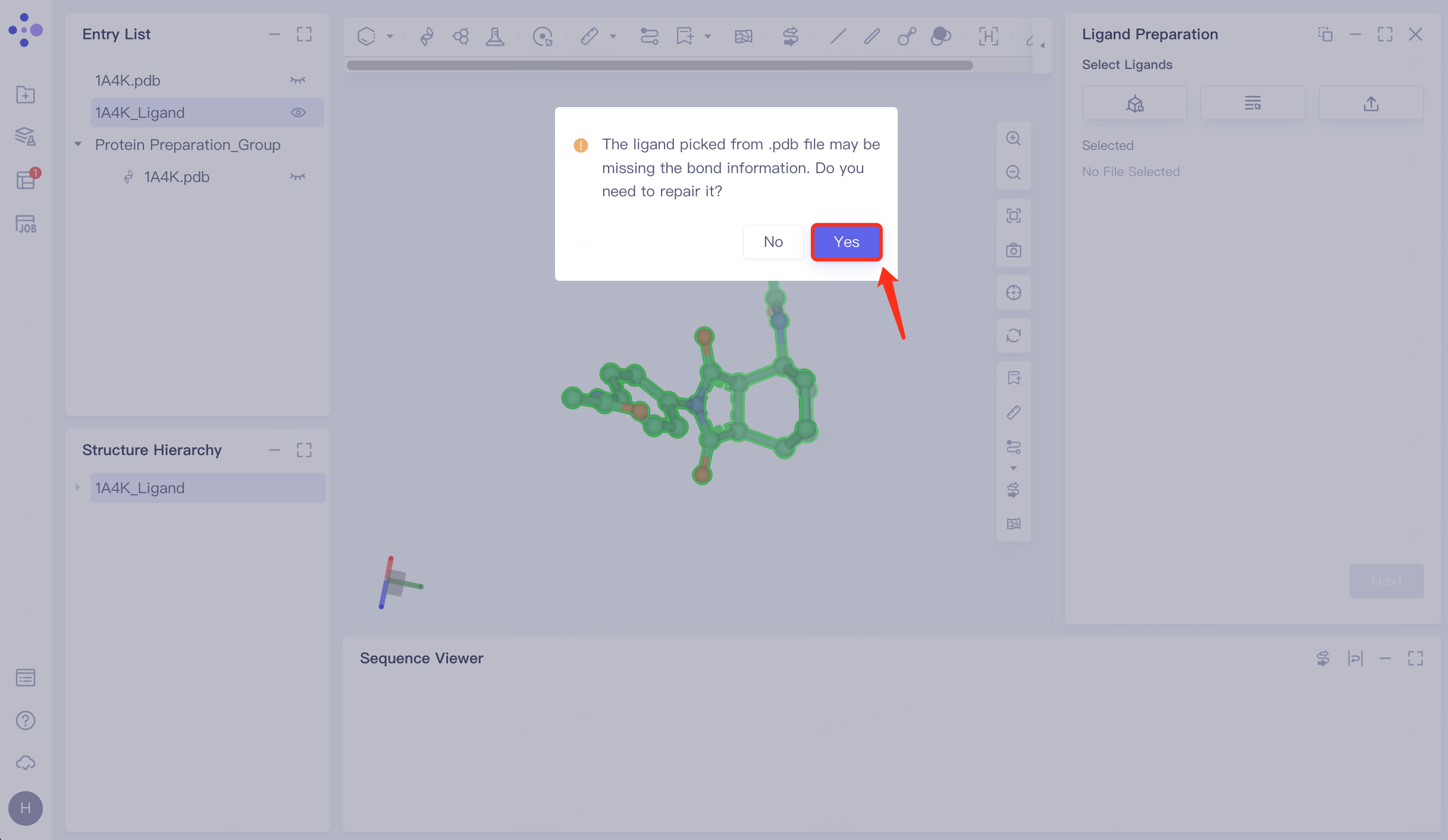
3.3 Fix missing keys
The prompt window pops up and click Yes to confirm the repair.
In the repaired ligand import task, click "Next" to go to the next step.
 |  |
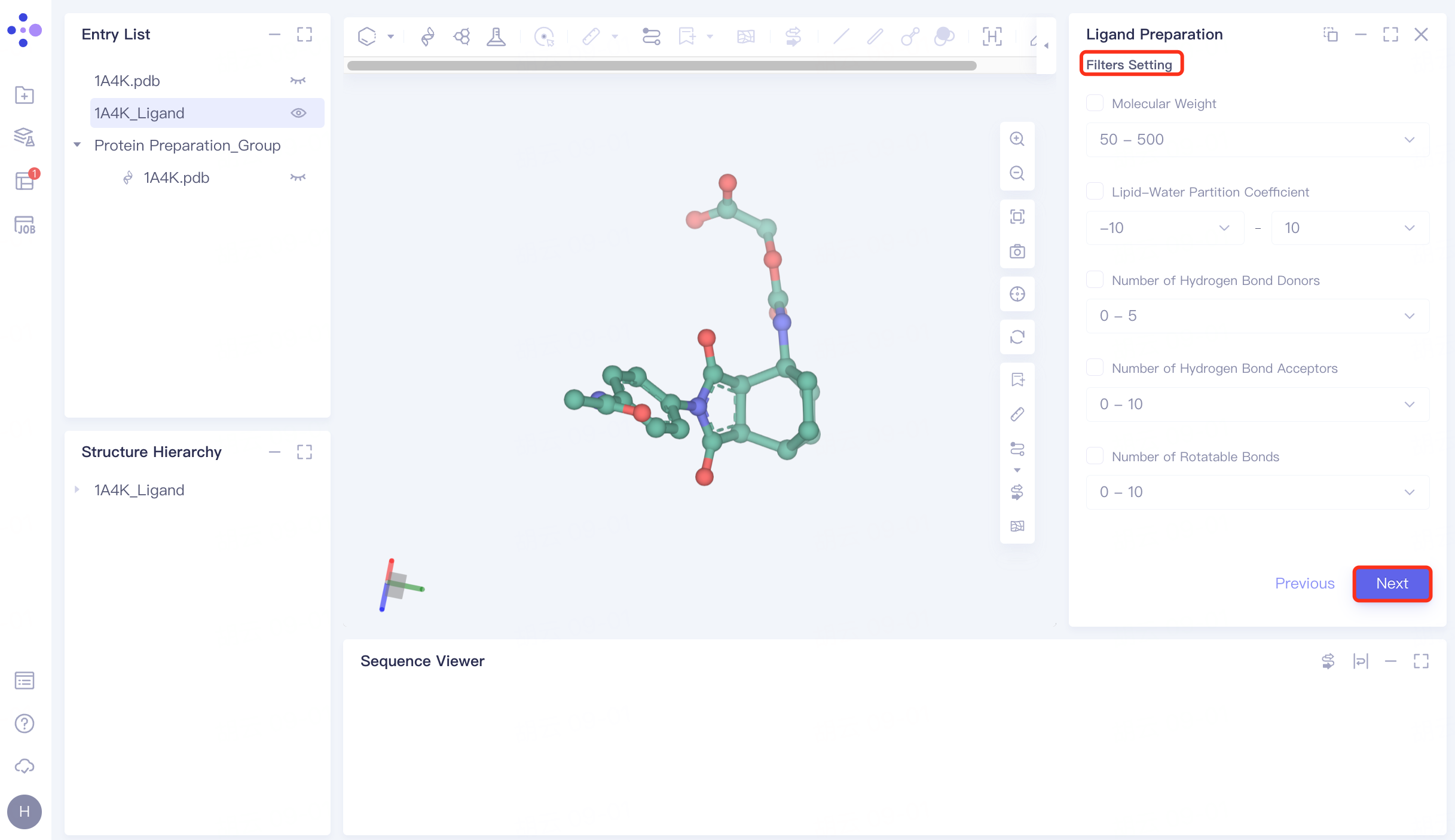
3.4 Filter Setting
No filter conditions are set here, click Next to skip this step.
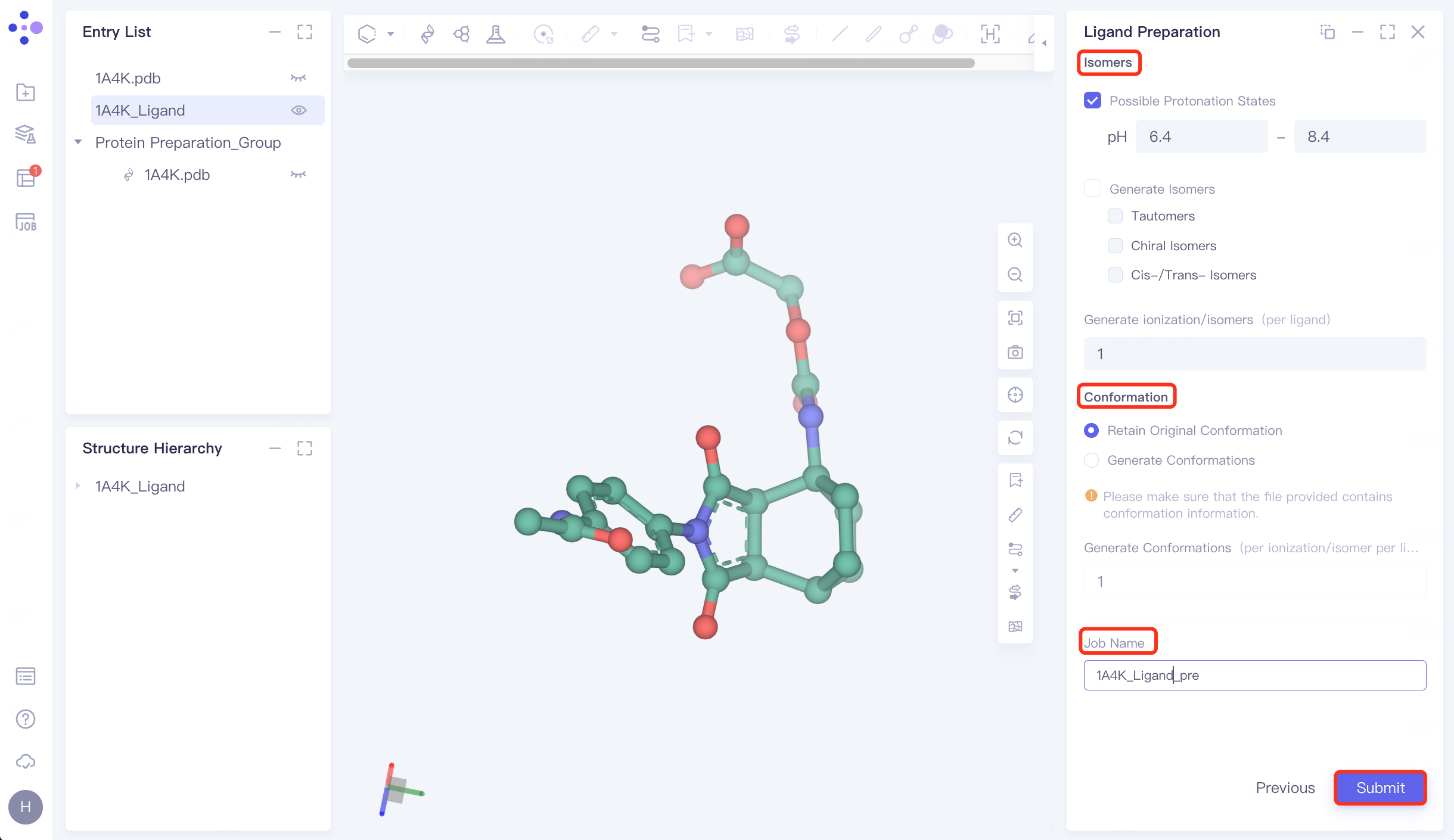
3.5 Generating Conformations
Isomers: Check Possible Protonation States and set the pH to 6.4~ 8.4;
Conformation: Select Retain Original Conformation to keep the original conformation;
Name the task "1A4K_Ligand_pre" at "Job Name";
Click "Submit" to submit the task.
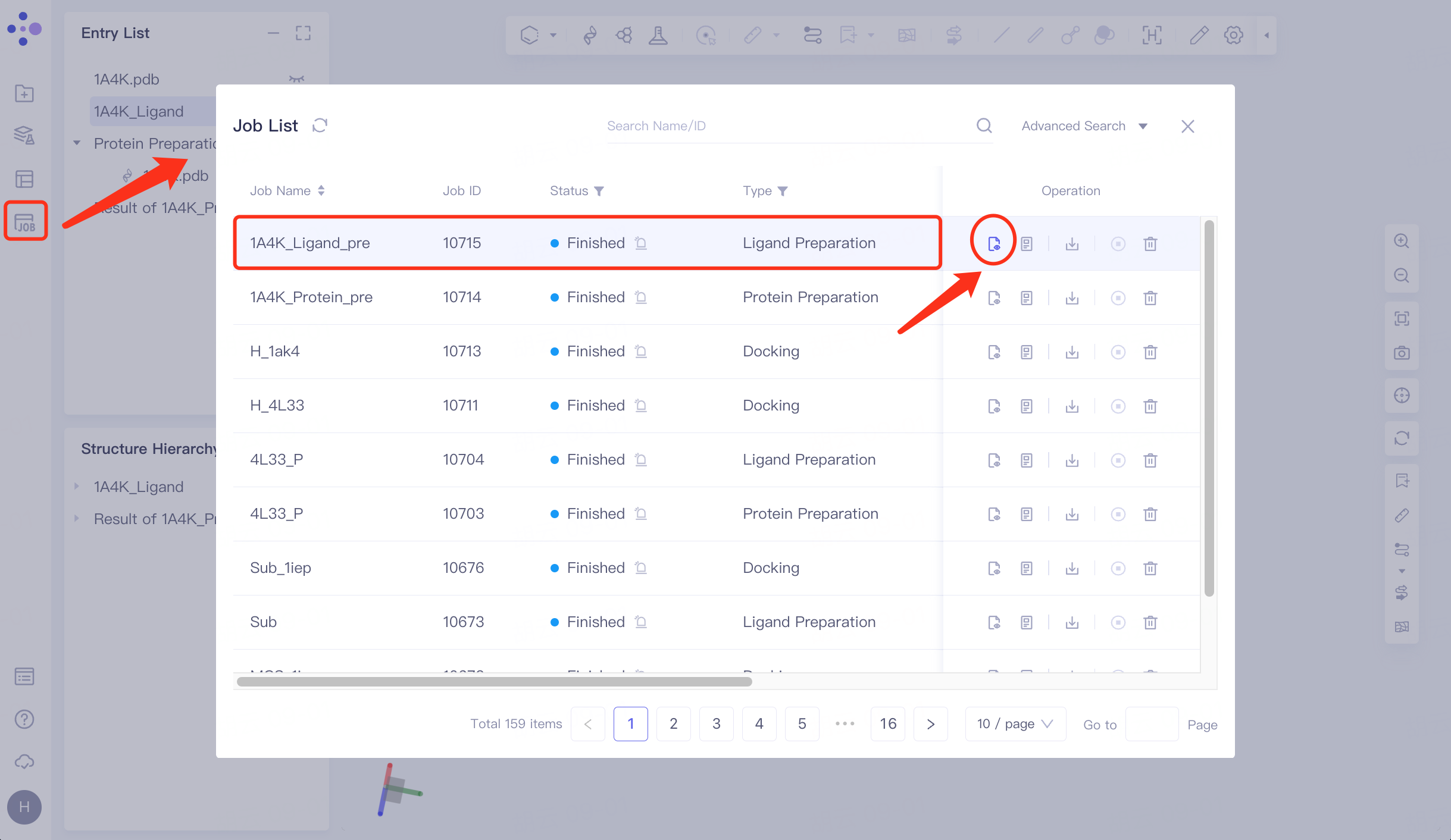
3.6 Check for ligands
Click Job in the left general menu bar → find the "1A4K_Ligand_pre" task in the pop-up Job List interface → click Show.
The right side pops up the results of ligand preparation.
 | 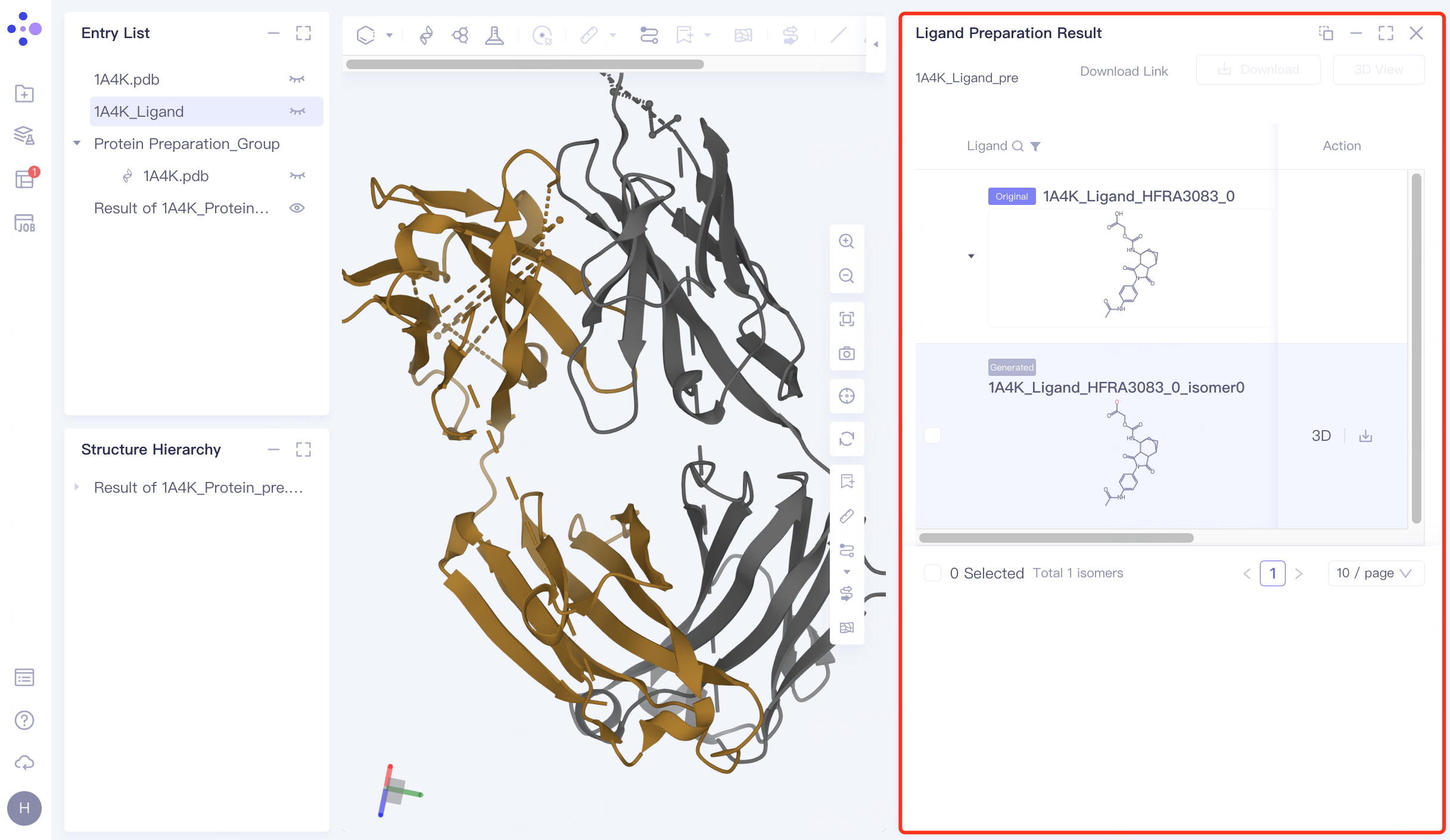 |
Click "3D" to display the prepared ligand in the 3D Workspace window to check the ligand structure.
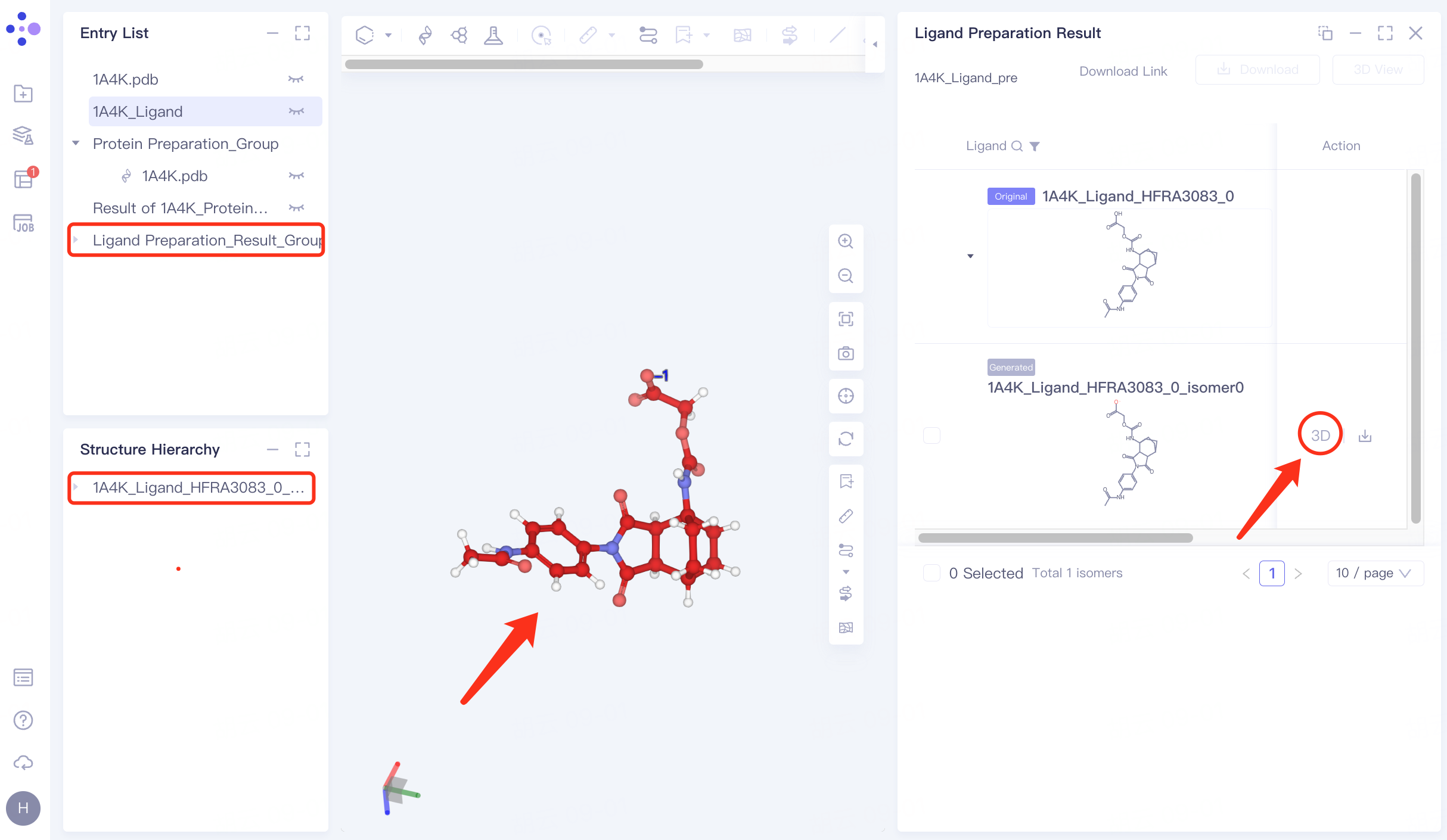
4. Docking
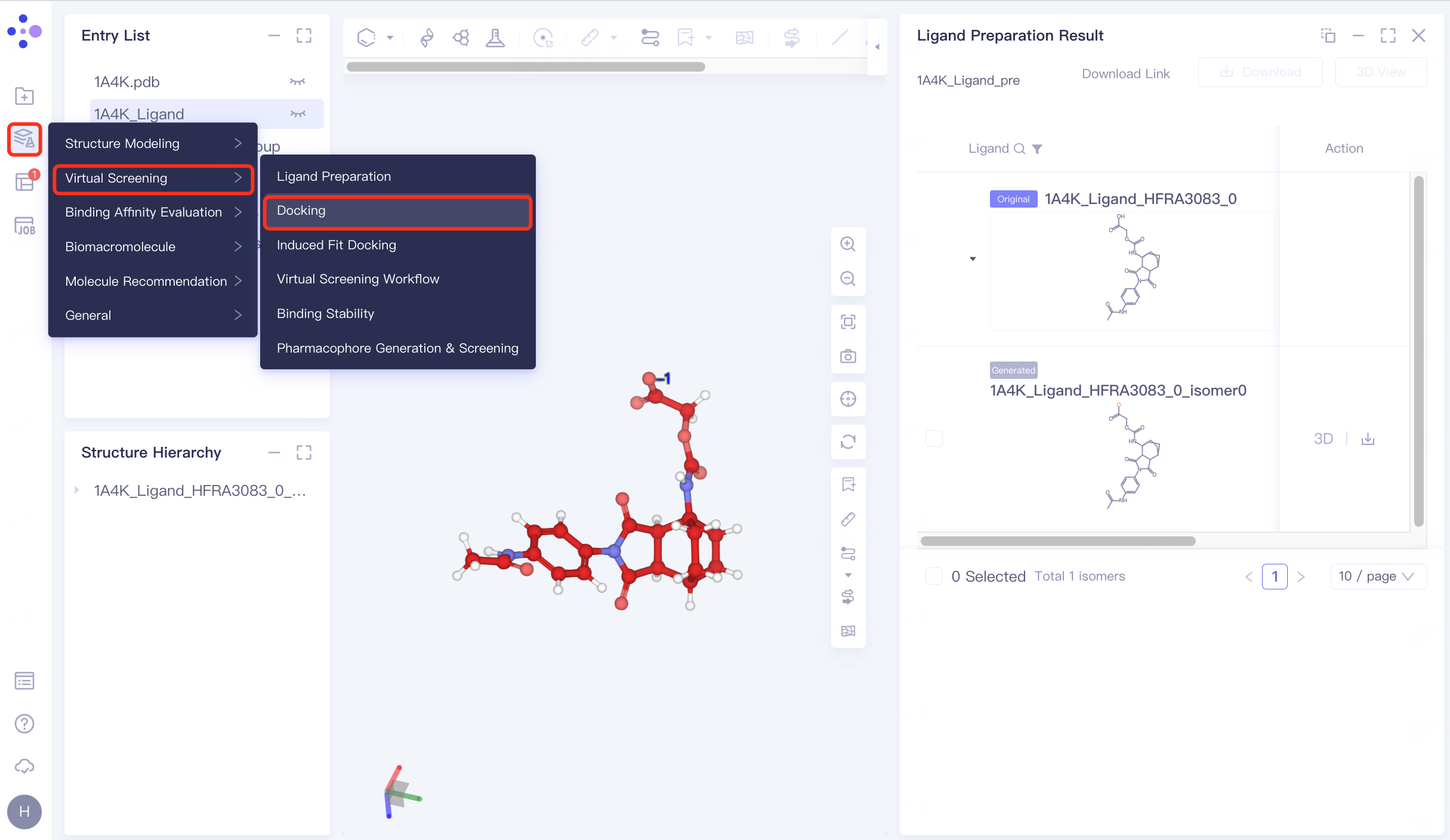
4.1 Entrance
The left general menu bar Function → Virtual Screening → Docking . The right interface appears.
 |  |
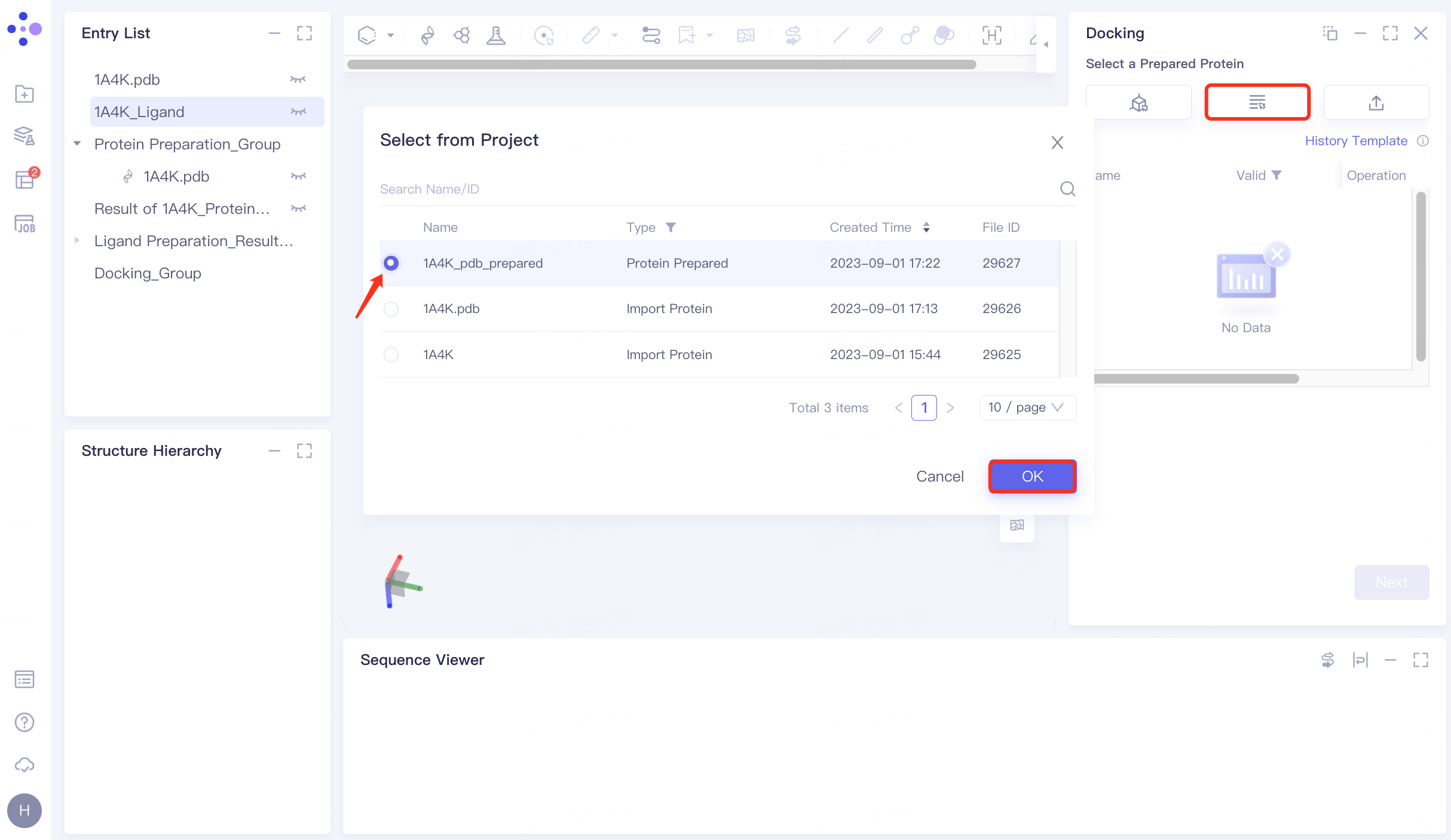
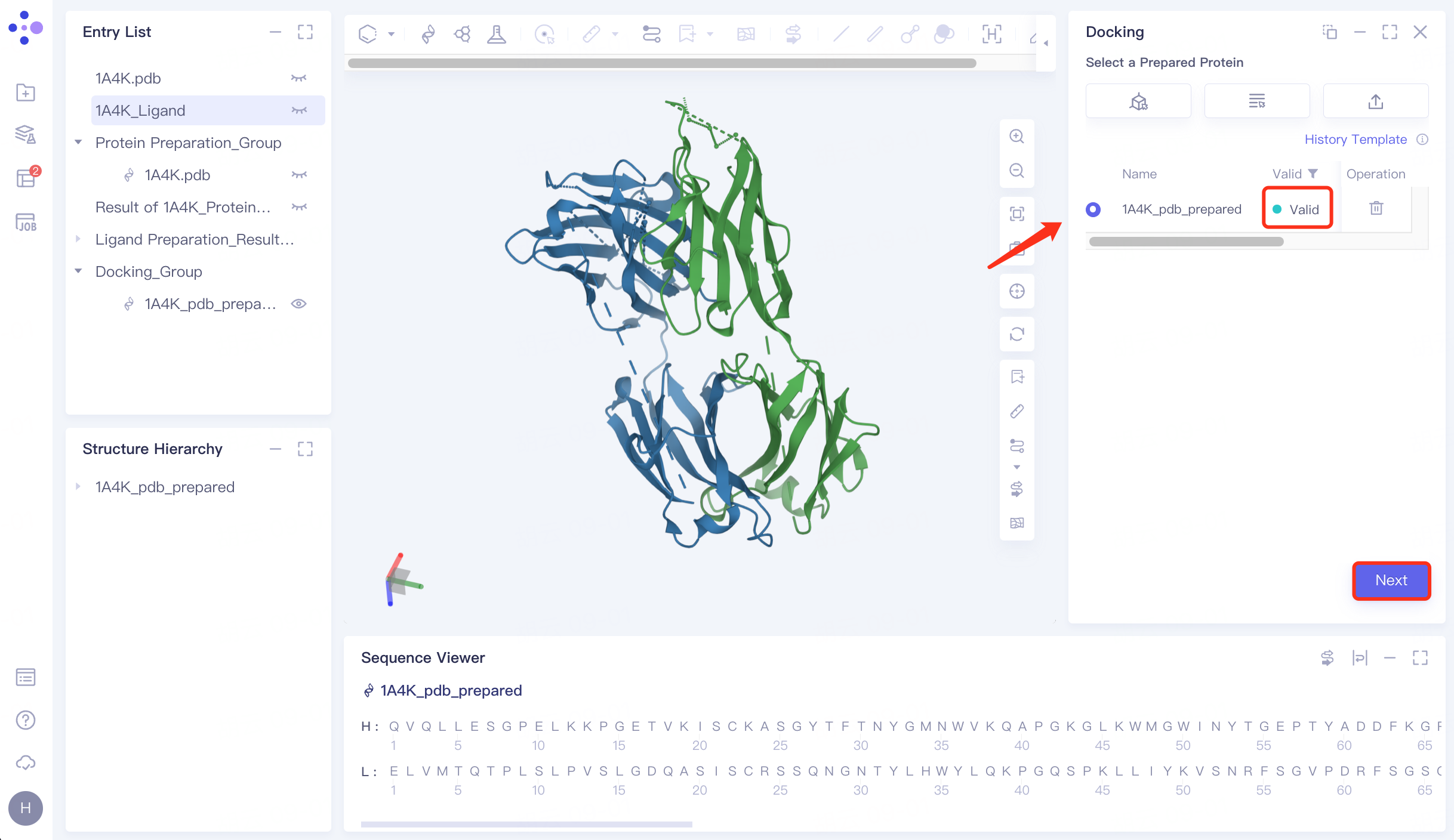
4.2 Upload receptor protein structure
Click "Select from Project" → Select "1A4K_pdb_prepared" in the pop-up window → Click "OK" → After passing the force field check, the status is displayed as "Valid" → Click "Next" to enter the next operation.
 |  |
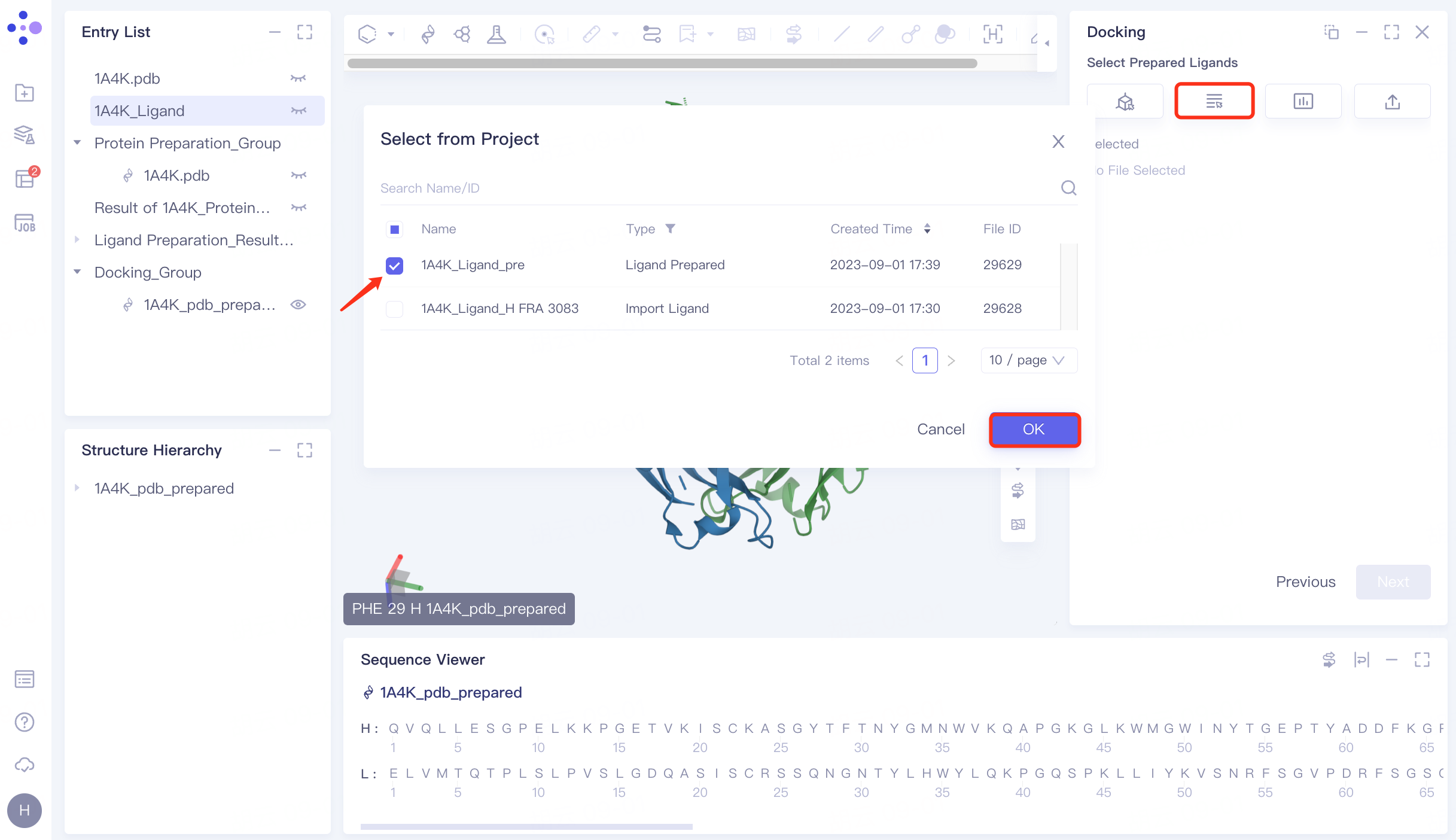
4.3 Upload ligand molecular structure
Click "Select from Project" → select "1A4K_Ligand_pre" → click "OK" in the pop-up window. After the molecule is uploaded successfully, click "Next" to proceed to the next step.
 |  |
4.4 Combined with pocket settings
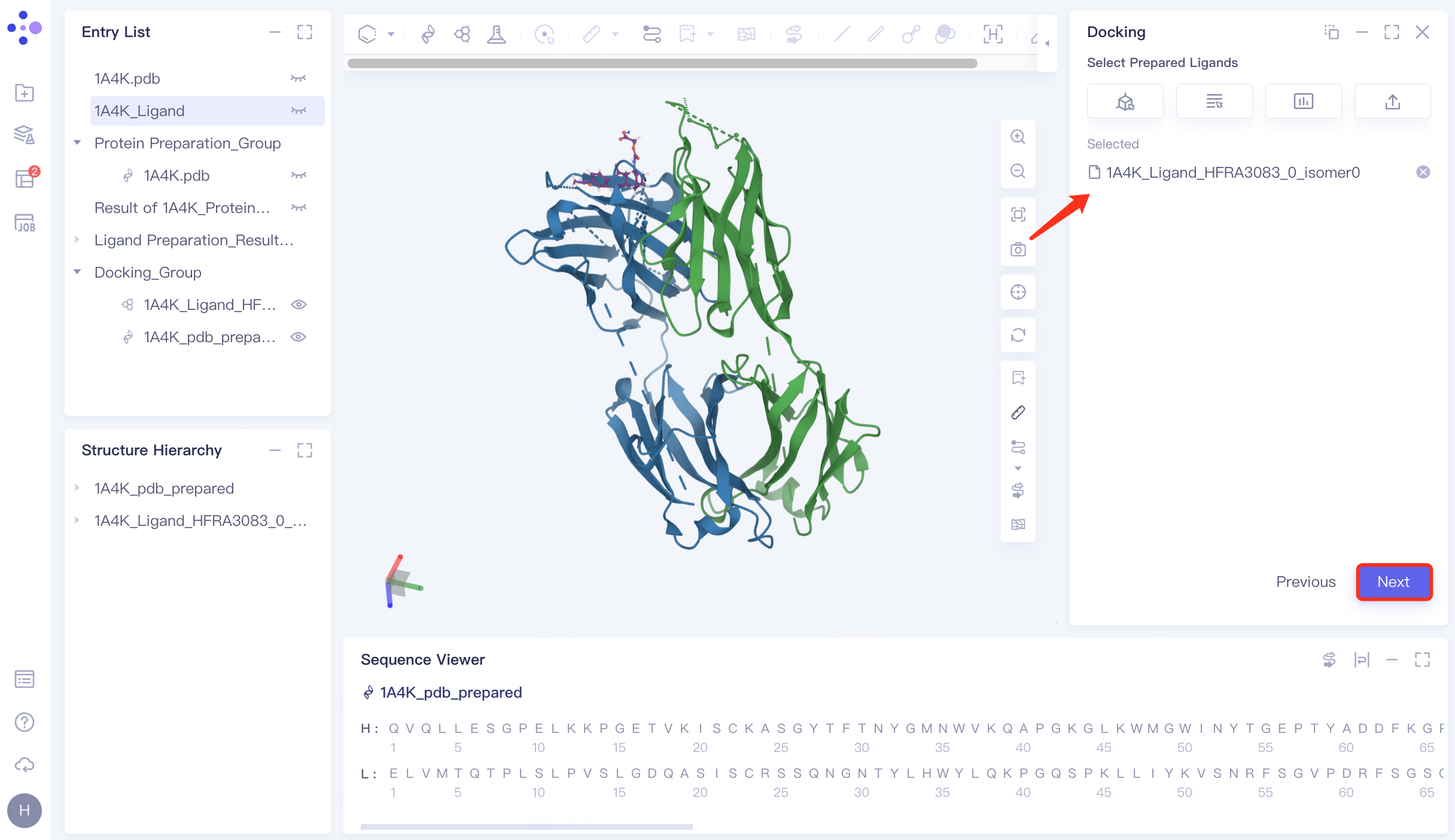
Display the "1A4K_Ligand" in the 3D workspace in the Entry List window → "Pocket From" in the "Pocket Setting" interface select "Select Ligand in the structure as center" → select "1A4K_Ligand H FRA 3083", which is the location of the "1A4K_Ligand" (the pocket where the ligand in the original eutectic is located) → Cutoff is set to 1.00 Å→ Click "Next" to proceed to the next step.
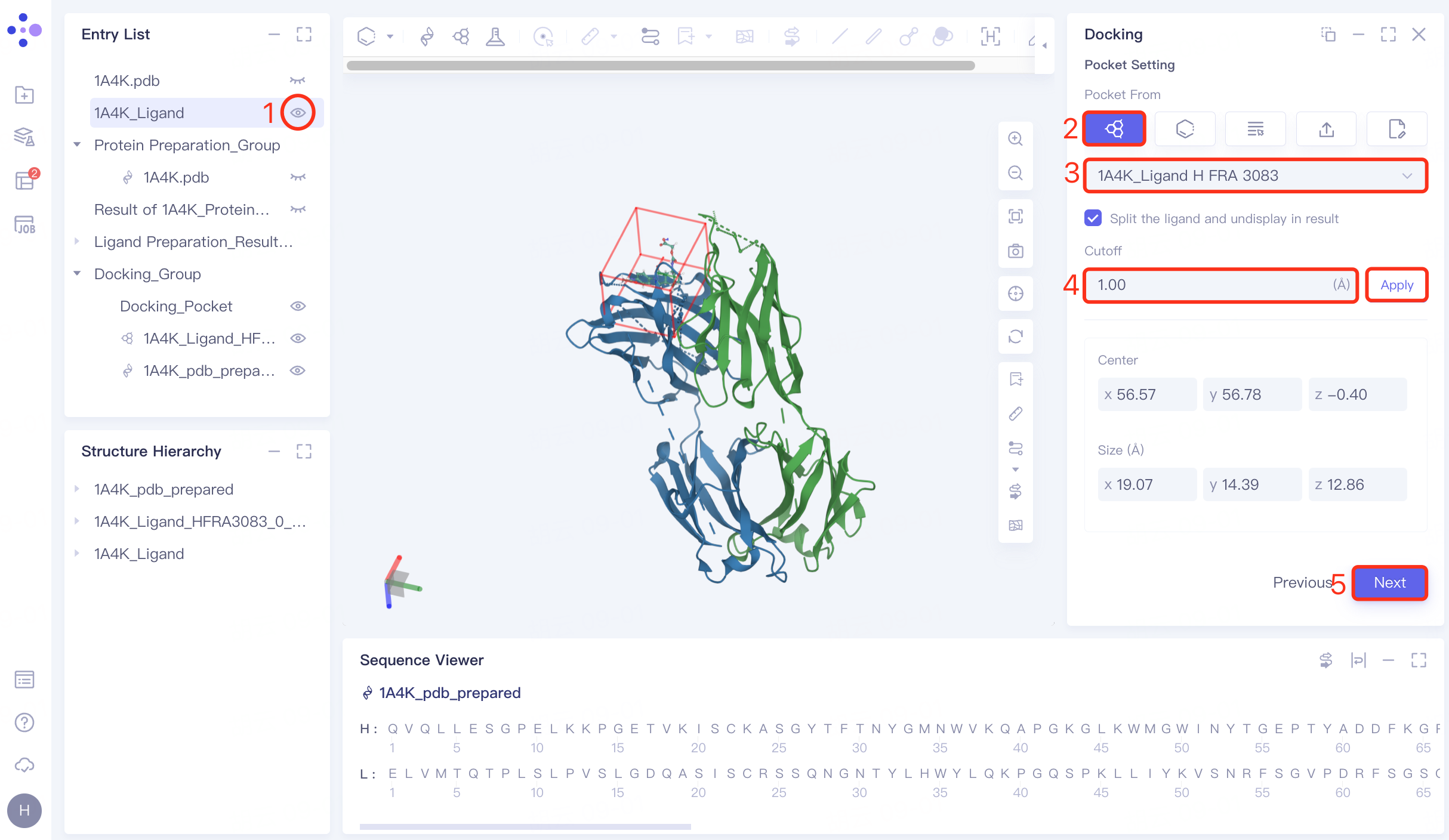
4.5 Constrained Docking
Pop up "Constrained Docking " interface → select "H-Bond" at "Type of Constrained Docking", and perform molecular docking based on hydrogen bond restriction → " Ligand Showed in 3D Workspace" select "1A4K_Ligand_HFRA3083_0_isomer0_AMOL" (in general, select the reference ligand that interacts with the receptor for hydrogen bonding, select the prepared ligand here) → click "Confirm".
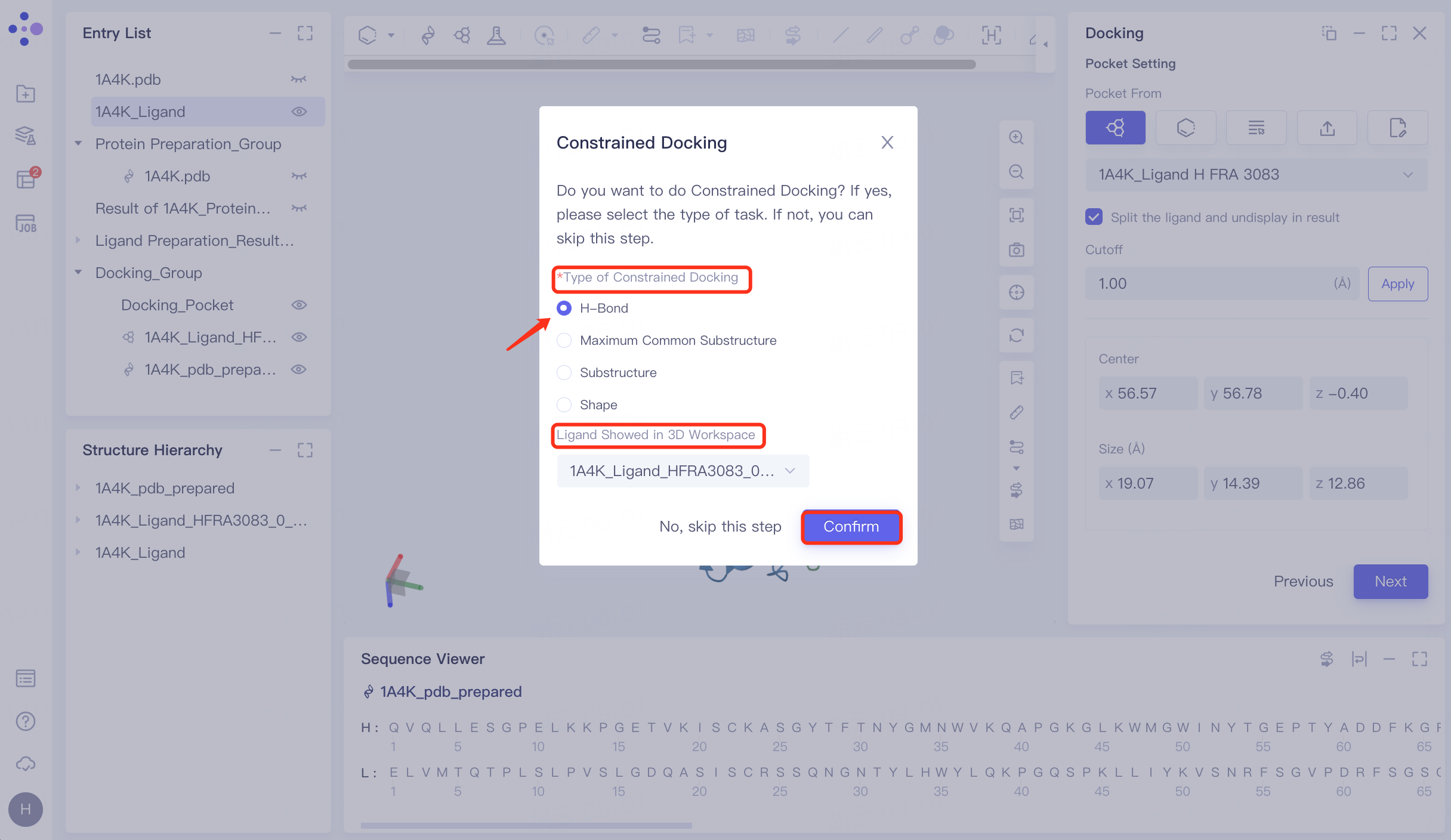
Protein residues within the Pocket are shown in the form of Lines, and the hydrogen bond interaction of the ligand with the receptor is represented by an orange dashed line.
In the 3D Workspace window, select the atom that interacts with the ligand . After selecting, the atom name will be automatically displayed in the Donors List and Acceptors List → click "Next" to enter the next step.
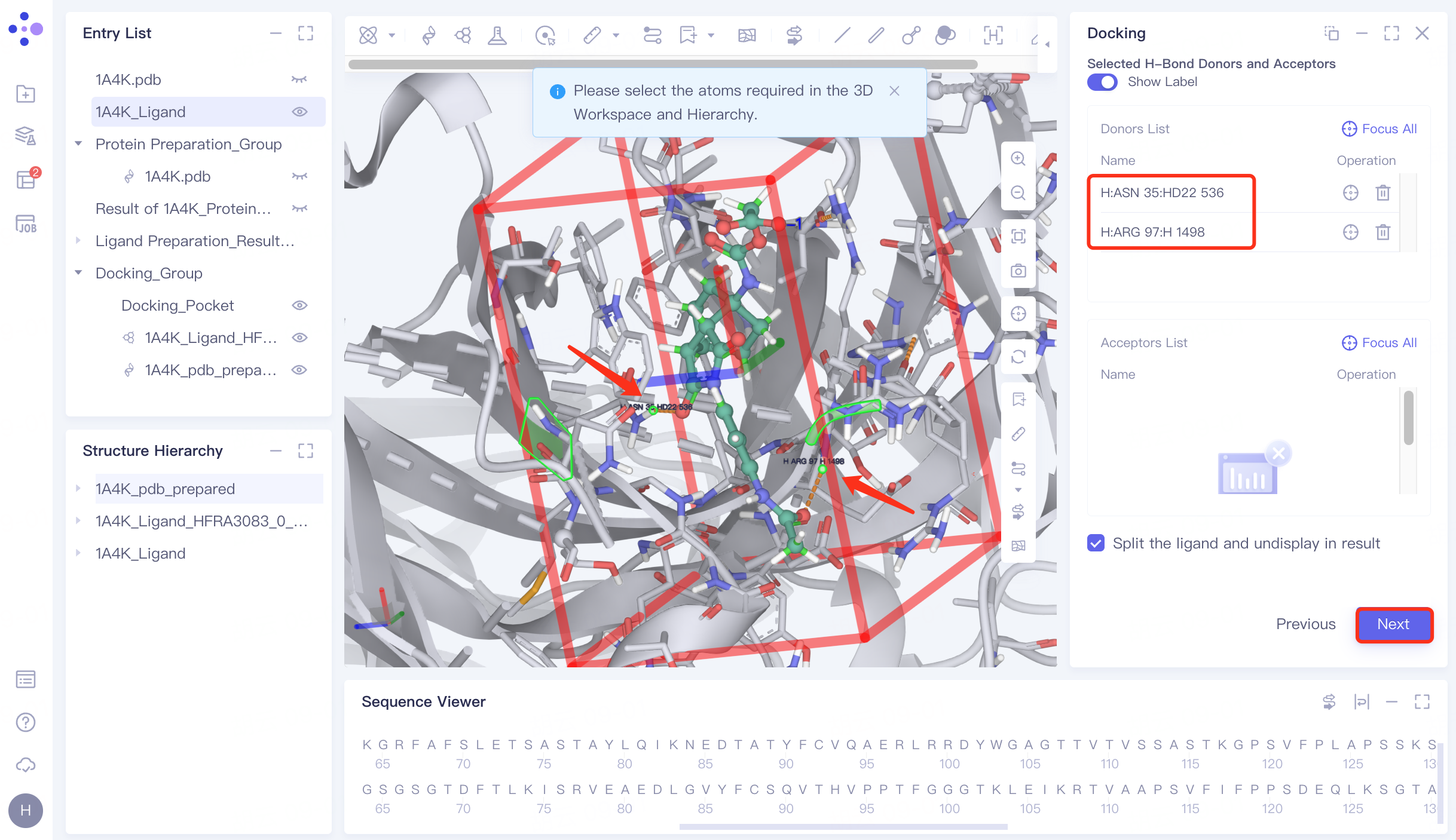
4.6 Docking parameter setting
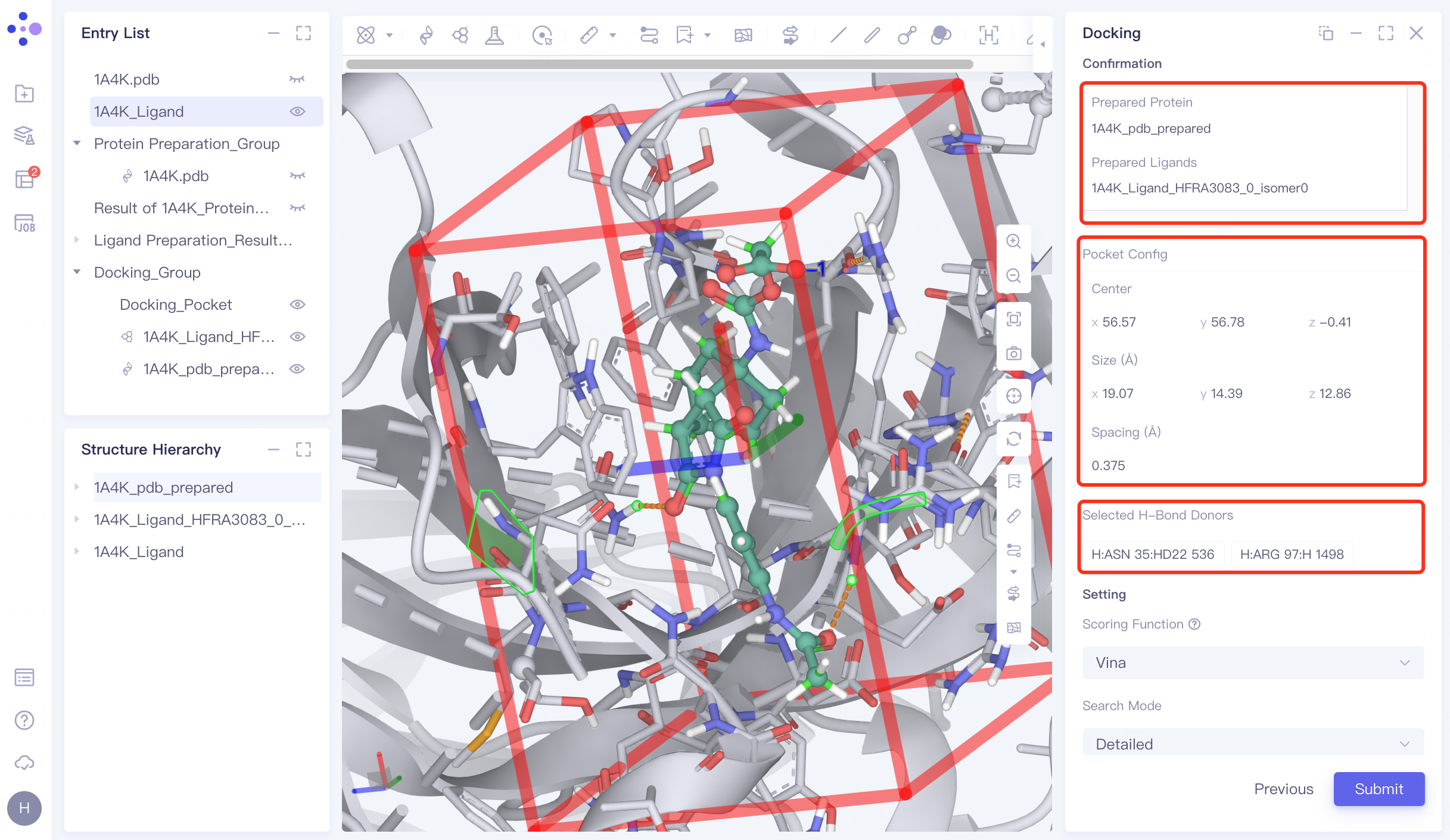
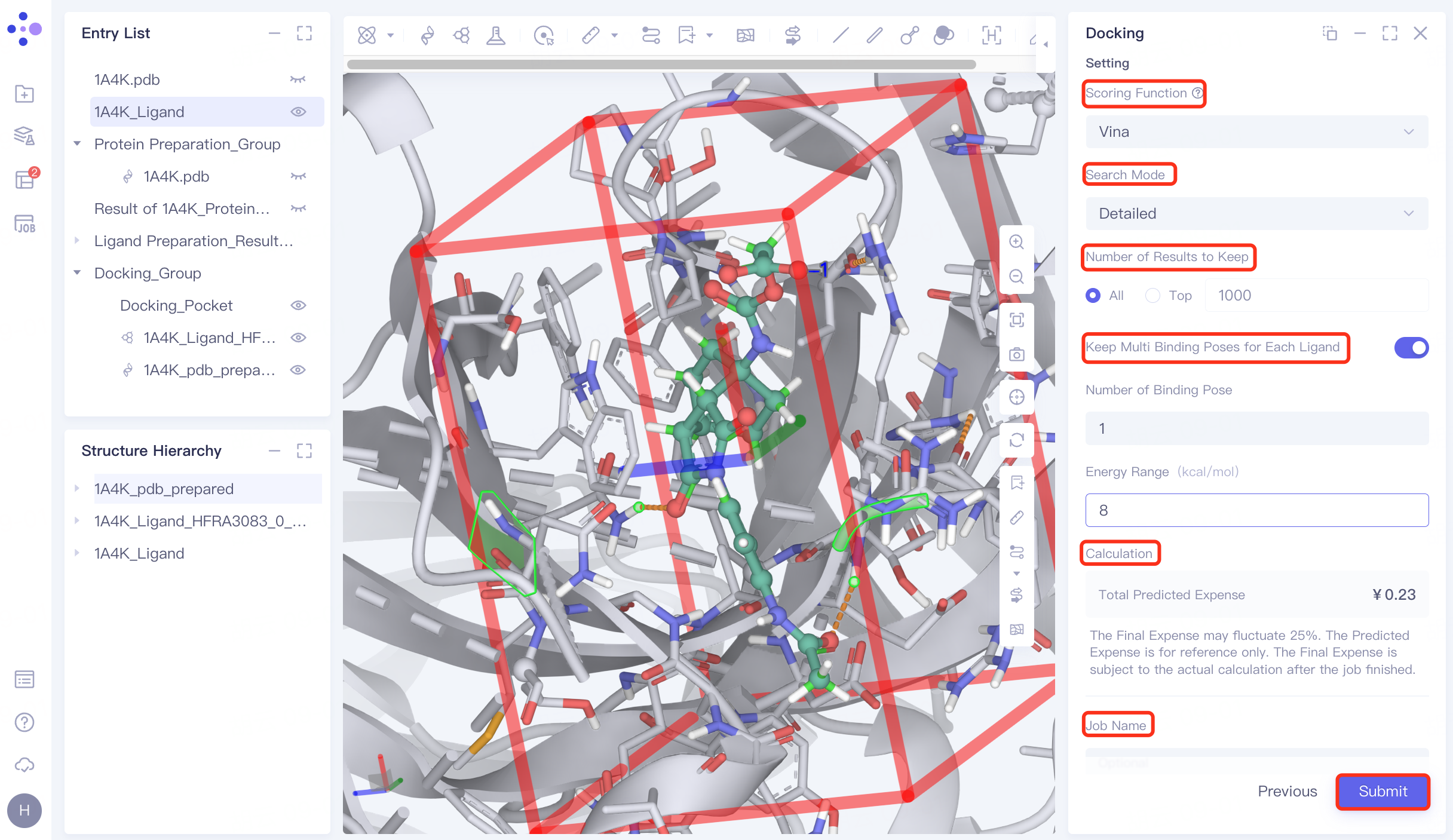
Confirmation and parameter setting: Confirm whether the receptor, ligand , pocket, and hydrogen bond check points are correct; Scoring Function (scoring function) selects Vina , Search Mode selects Detailed, Number of Results to Keep selects All, and open Keep Multi Binding Poses for Each Ligand , set Number of Binding Pose to 8, and Energy Range to 9. Confirm the calculation cost at Calculation, name the task "H_1A4K" in Job Name, and click "Submit" to submit the task.
 |  |
5. Results analysis
5.1 Display of results
Click Job in the left general menu bar → find the "H_1A4K" task in the pop-up Job List interface → click Show.
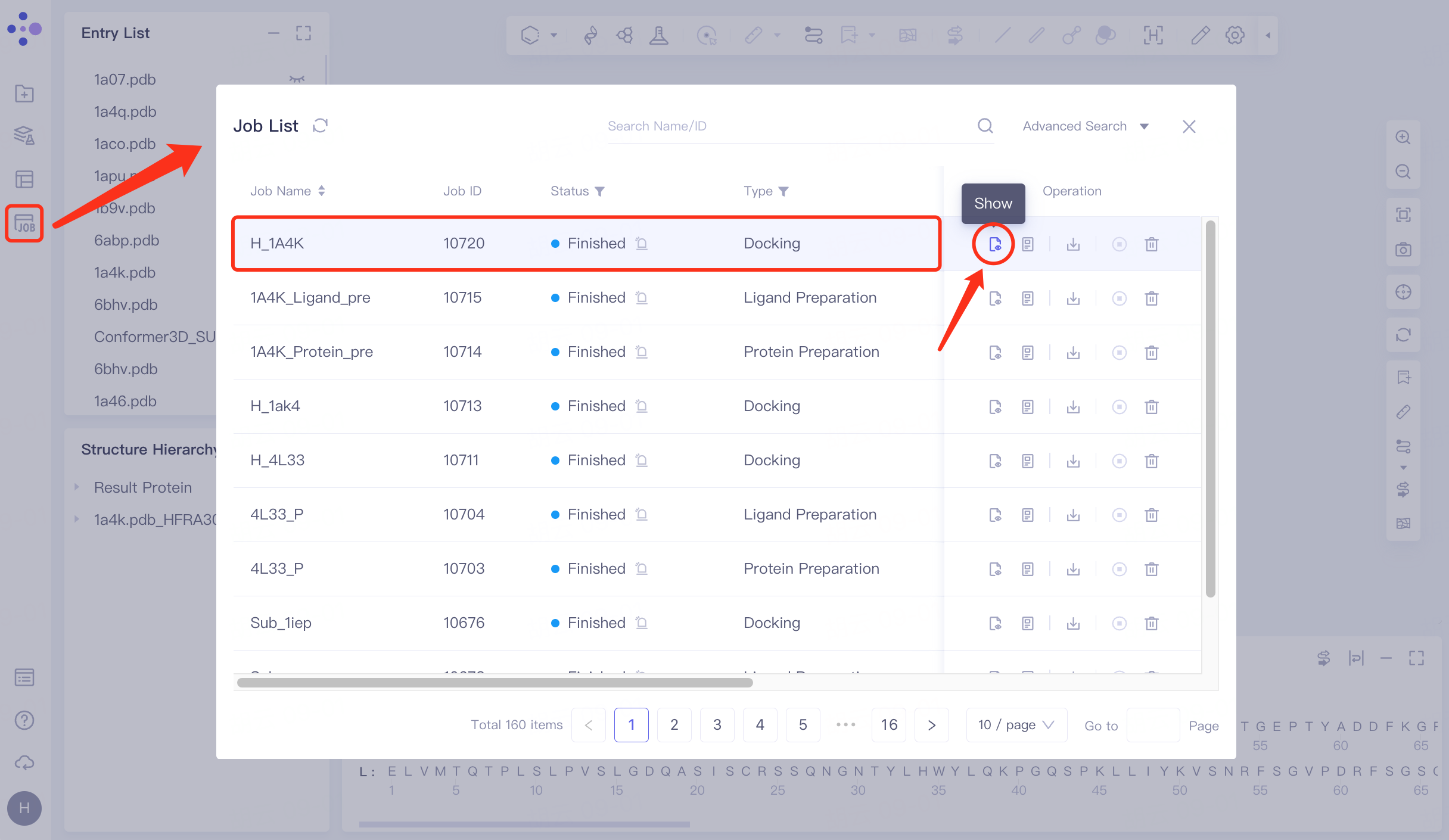
Open the interface as follows:
The docking results are sorted by "Constrained Score" by default, and the lowest "Constrained Score" is observed to be -11.19.
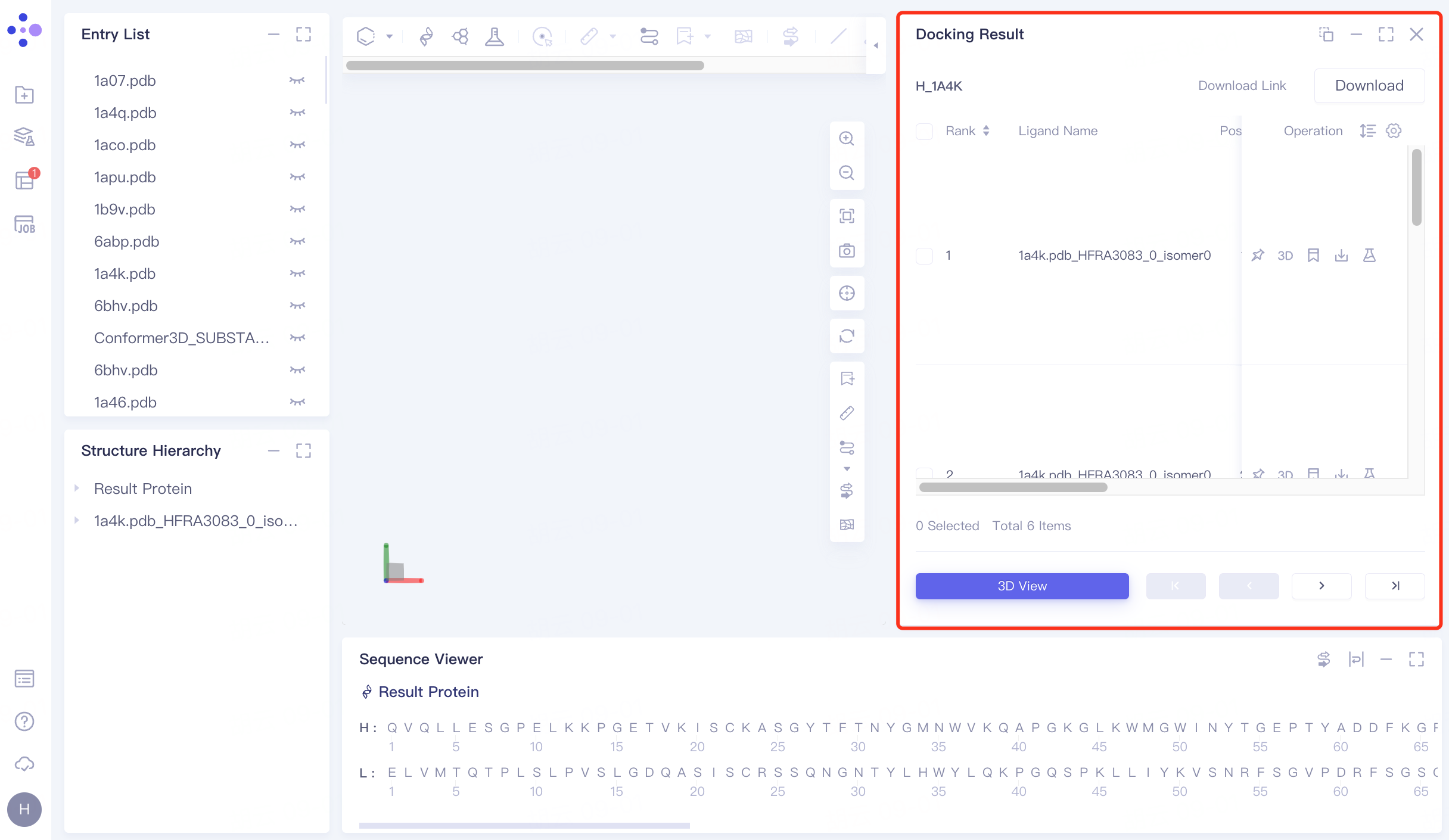
For the ligand with the lowest "Constrained Score", click 3D under Operation on the right to display the docking result of the ligand molecule with the receptor in the 3D Workspace window.
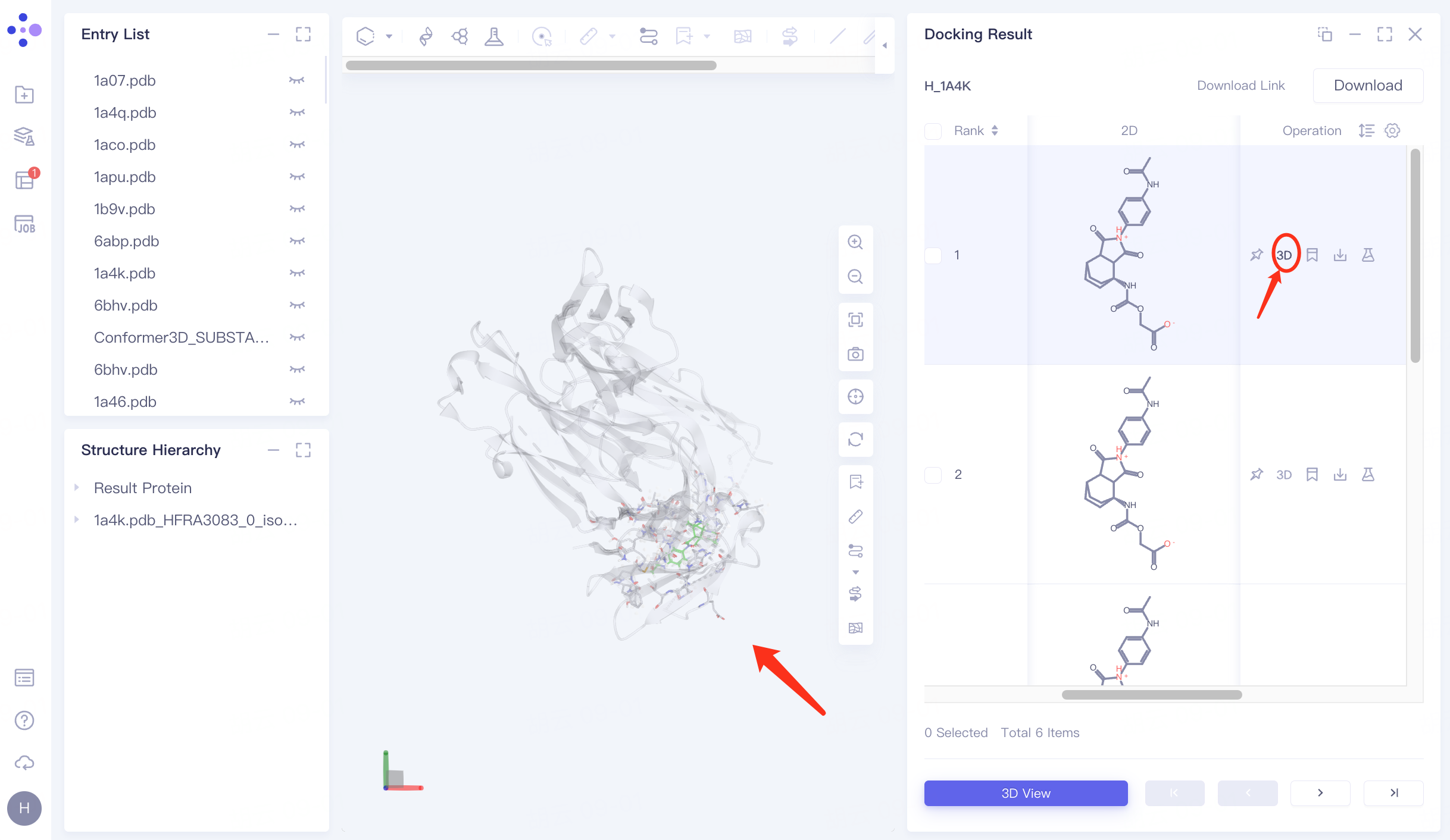
5.2 Hydrogen Bond Interaction View
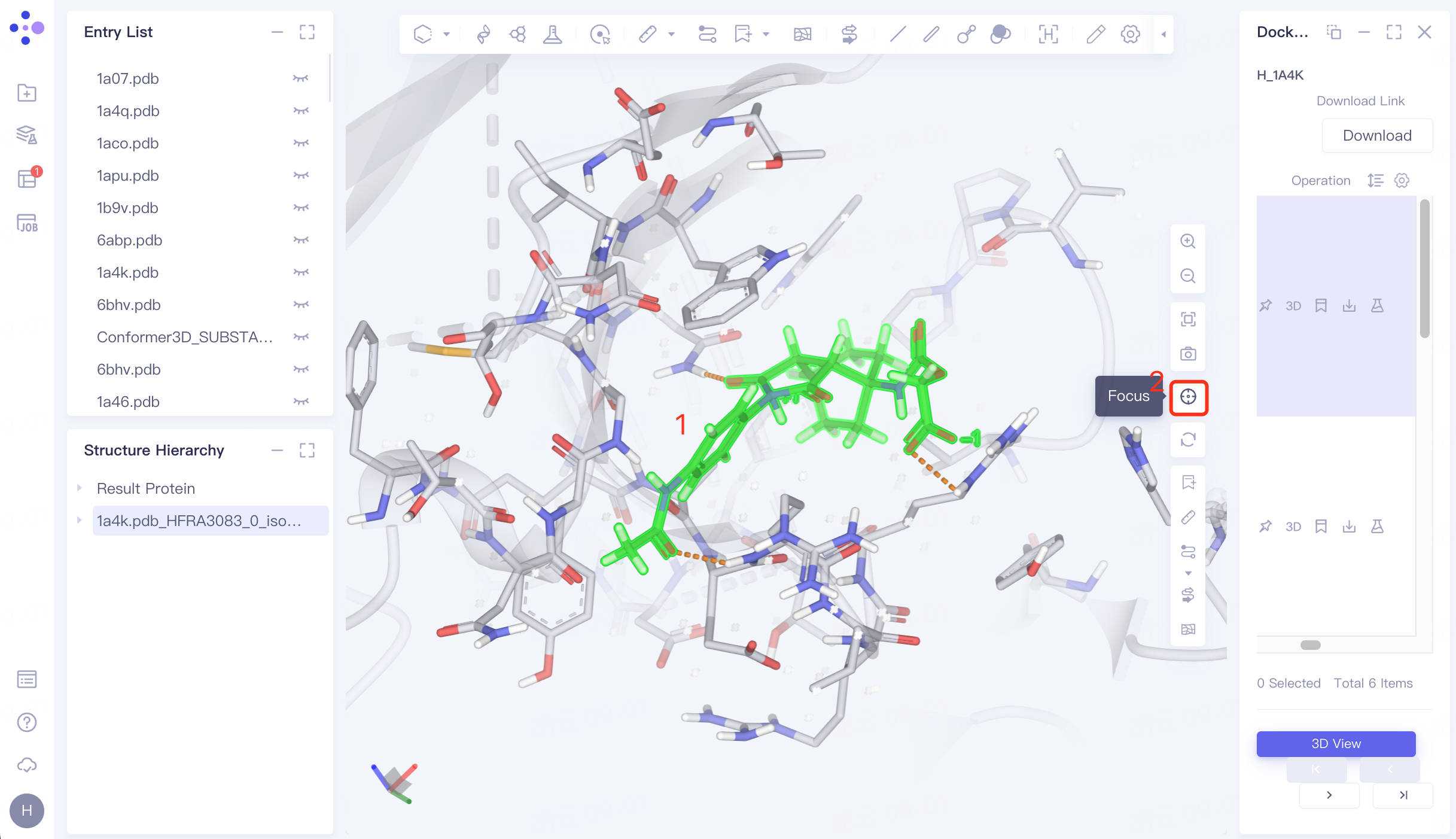
Select the docked ligand in the 3D Workspace window → click "Focus" → mouse wheel to zoom in on the ligand.
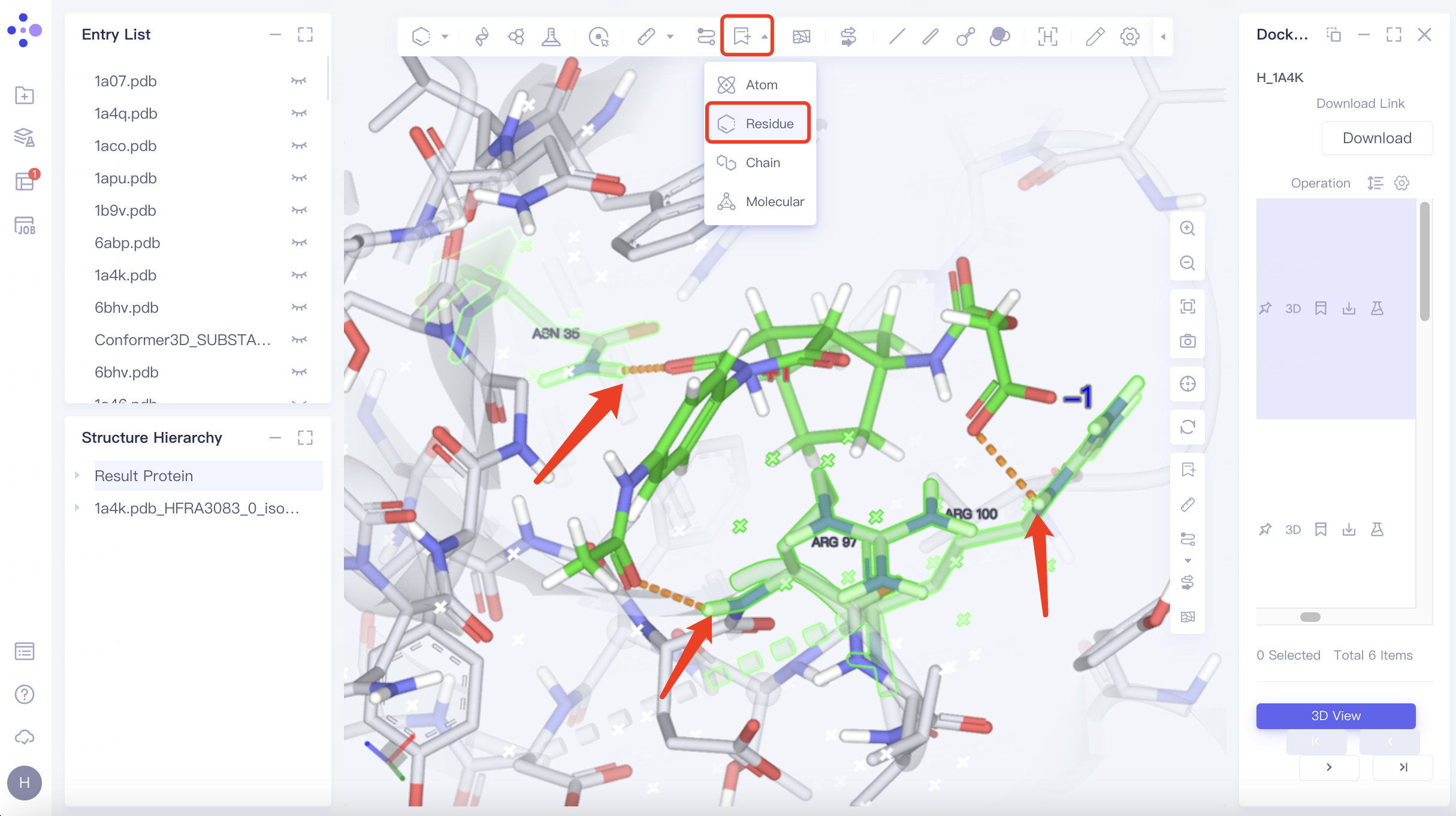
Adjust "Pick" to " Residue " in the toolbar at the top left of the 3D Workspace window → Select the residue that interacts with the ligand → Click "Label" in the toolbar at the top of the 3D Workspace window, select "Residue", and mark the name of the selected residue.
As shown in the figure, the docked ligand has hydrogen bonding interactions with the receptor residues "ASN 35", "ARG 97" and "ARG 100", which match the hydrogen bonding interactions between the receptor ligands in the eutectic.
5.3 Ligand pose accuracy check
The original Ligand is adjusted to the display state in the Entry List window. The original Ligand is displayed in orange in the 3D Workspace window, and the docked Ligand is displayed in green.
Hide Protein: Adjust "Pick" to "Molecular" in the toolbar above the 3D Workspac window → Select two chains of protein → Click "Ribbon" in the upper toolbar to hide the secondary structure of the protein → Click "Line" in the upper toolbar to hide the residues expressed in Line form. The adjusted visual interface is shown in the figure.
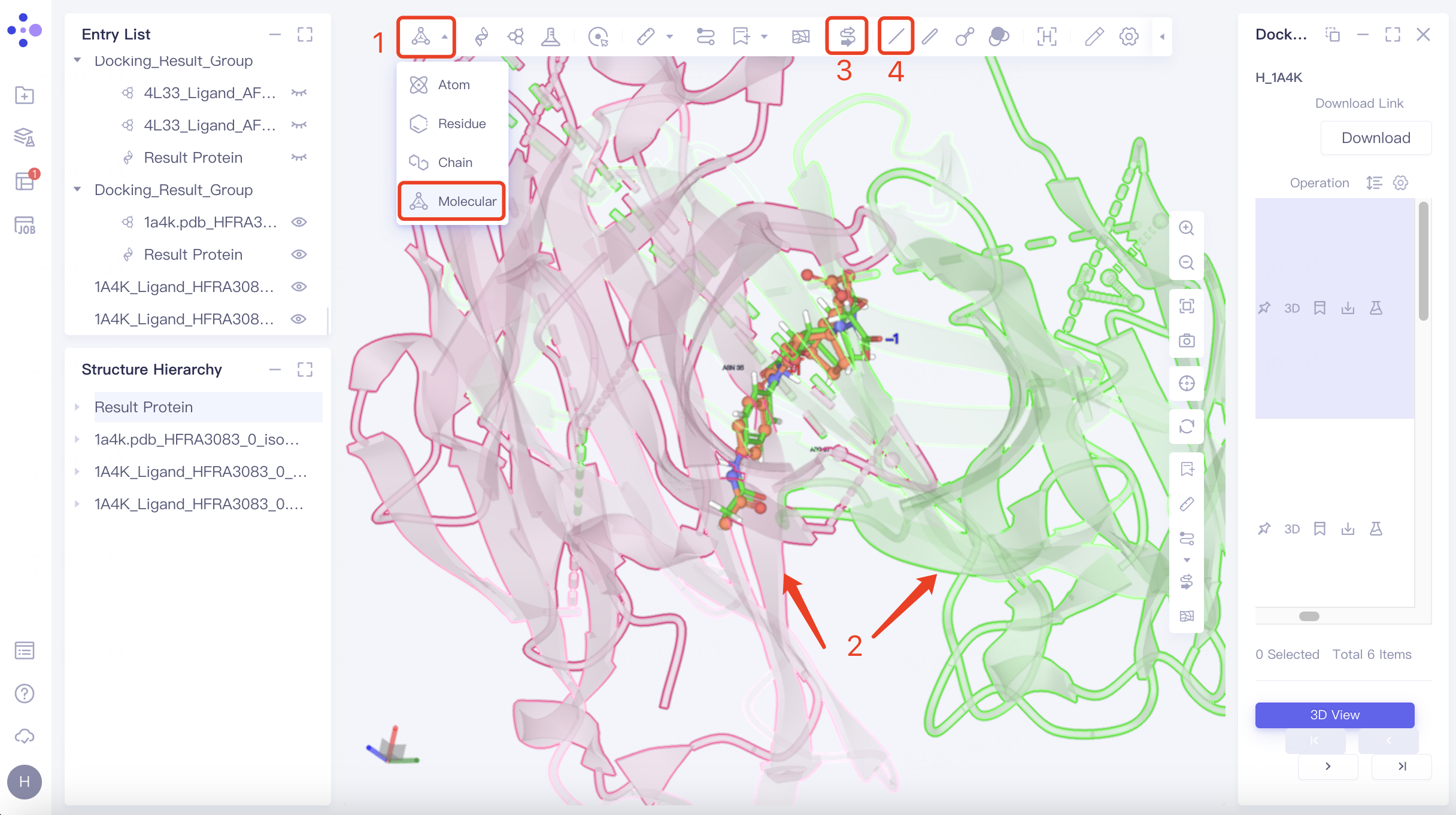 | 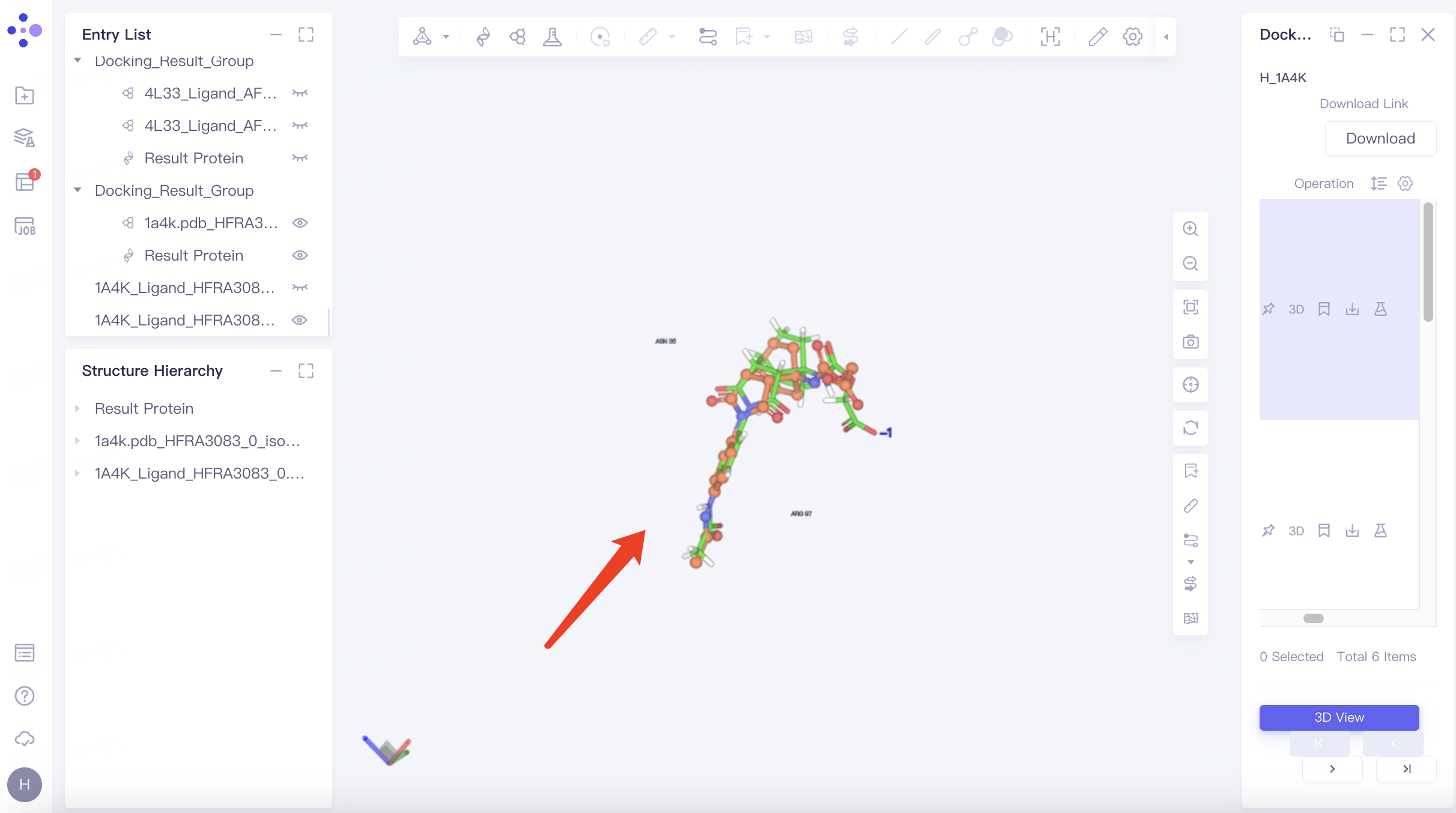 |
Zoom in on ligand: Select any ligand → click "Reset" in the toolbar on the right side of the 3D Workspac window → push the mouse wheel to zoom in on the ligand.
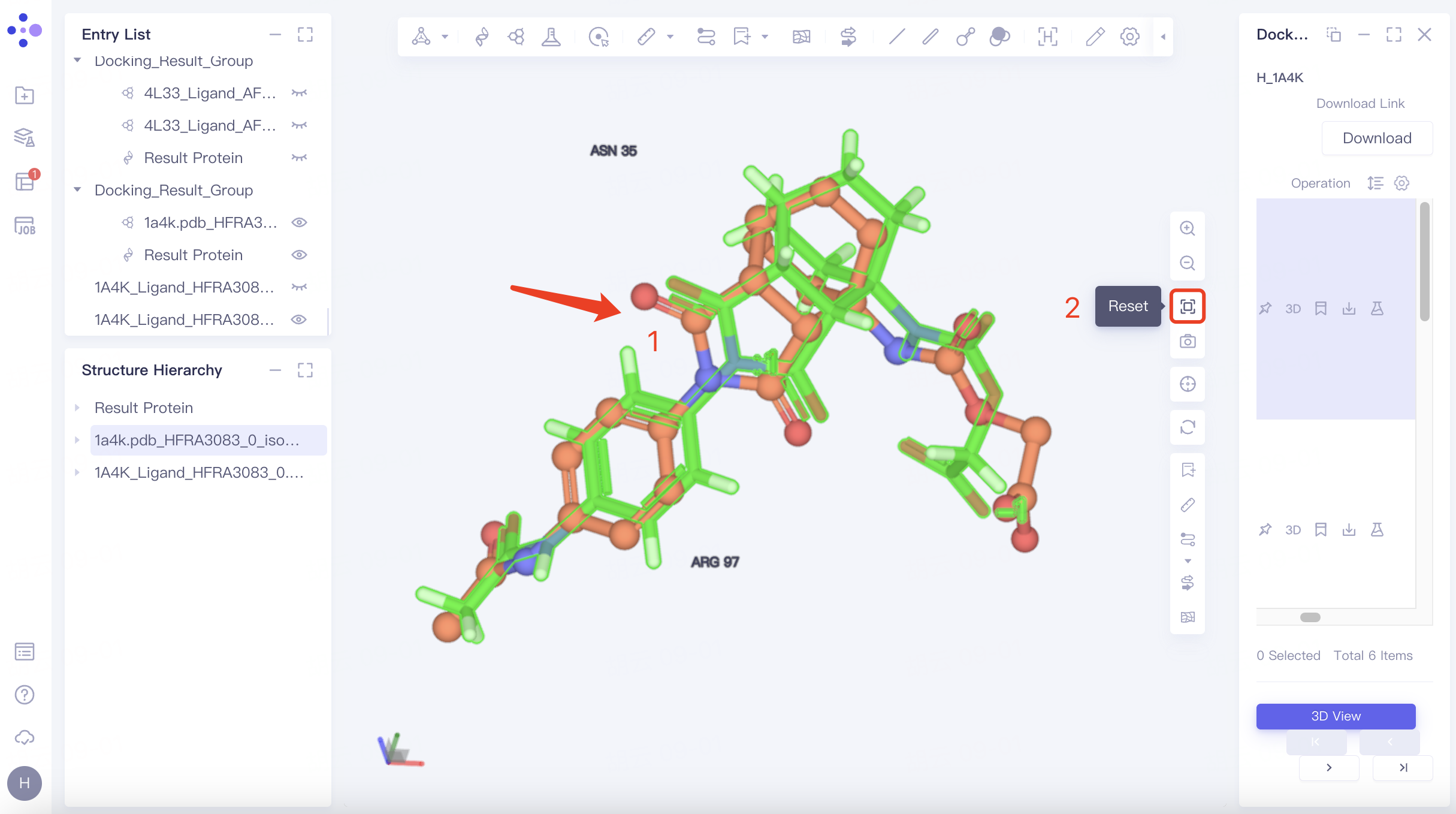
From the visualization window, it can be seen that the difference between the docked ligand pose and the original pose is small, and the pose is more reasonable.
6. References
[1] Romesberg F E, Spiller B, Schultz P G, et al. Immunological origins of binding and catalysis in a Diels-Alderase antibody[J]. Science, 1998, 279(5358): 1929-1933.