Virtual Screening Workflow
1. 前言
使用分子对接进行快速的虚拟筛选,是小分子药物开发过程中常见的技术手段。Hermite®平台的Virtual Screening Workflw模块提供了高通量虚拟筛选功能。
基于学术界和工业界广泛认可的Docking引擎,深势科技的开发人员进行了GPU并行计算的深度优化,结合Hermite平台内置的海量虚拟化合物库,您可以快速完成大化学空间的虚拟筛选任务。
2. 使用方法
2.1 入口
- 左侧通用菜单栏Function → Virtual Screening → Virtual Screening Workflow。
- 右侧出现Virtual Screening Workflow窗口(红框内所示),整体界面如下:
2.2 操作
2.2.1 蛋白准备
-
蛋白结构输入,共有四种方式:
-
1)Select from 3D Workspace:点击Select from 3D Workspace选框 → 弹出Select Structure界面 → 界面左侧Structure Hierarchy/Sequence Viewer/3D Workspace 窗口选择所需蛋白结构 → Select Structure框内显示选中的蛋白名称,点击OK。

- 2)Select from Project:点击Select from Project选框 → 界面中间弹出Select from Project窗口,显示历史项目中的蛋白结构 → 选中所需蛋白结构 → 点击OK。

- 3)Select File:从本地文件夹中选择所需蛋白结构(.pdb格式)并上��传。

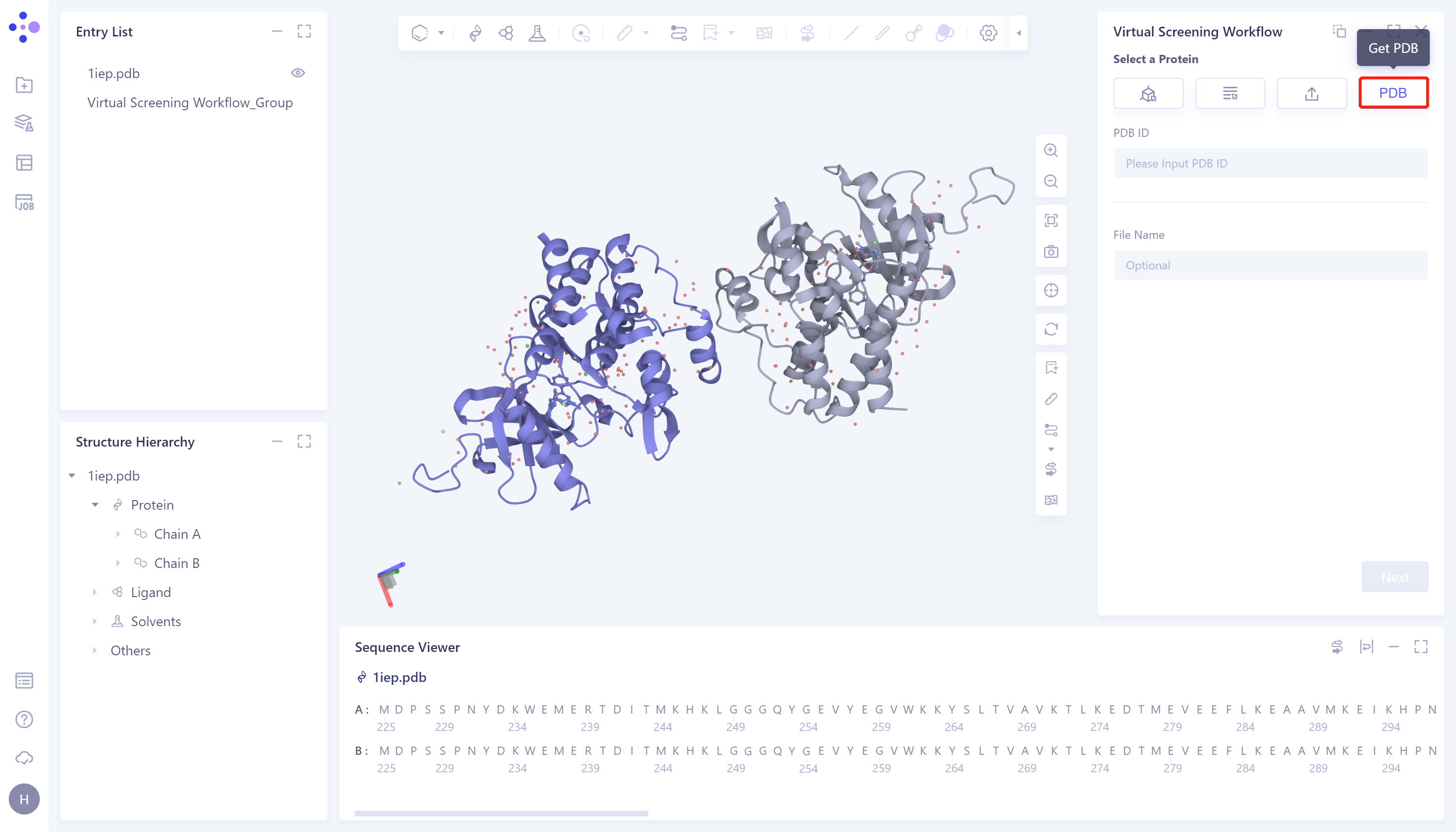
- 4)Get PDB:根据蛋白PDB ID从PDB数据库中检索并导入蛋白结构。
-
蛋白处理:
-
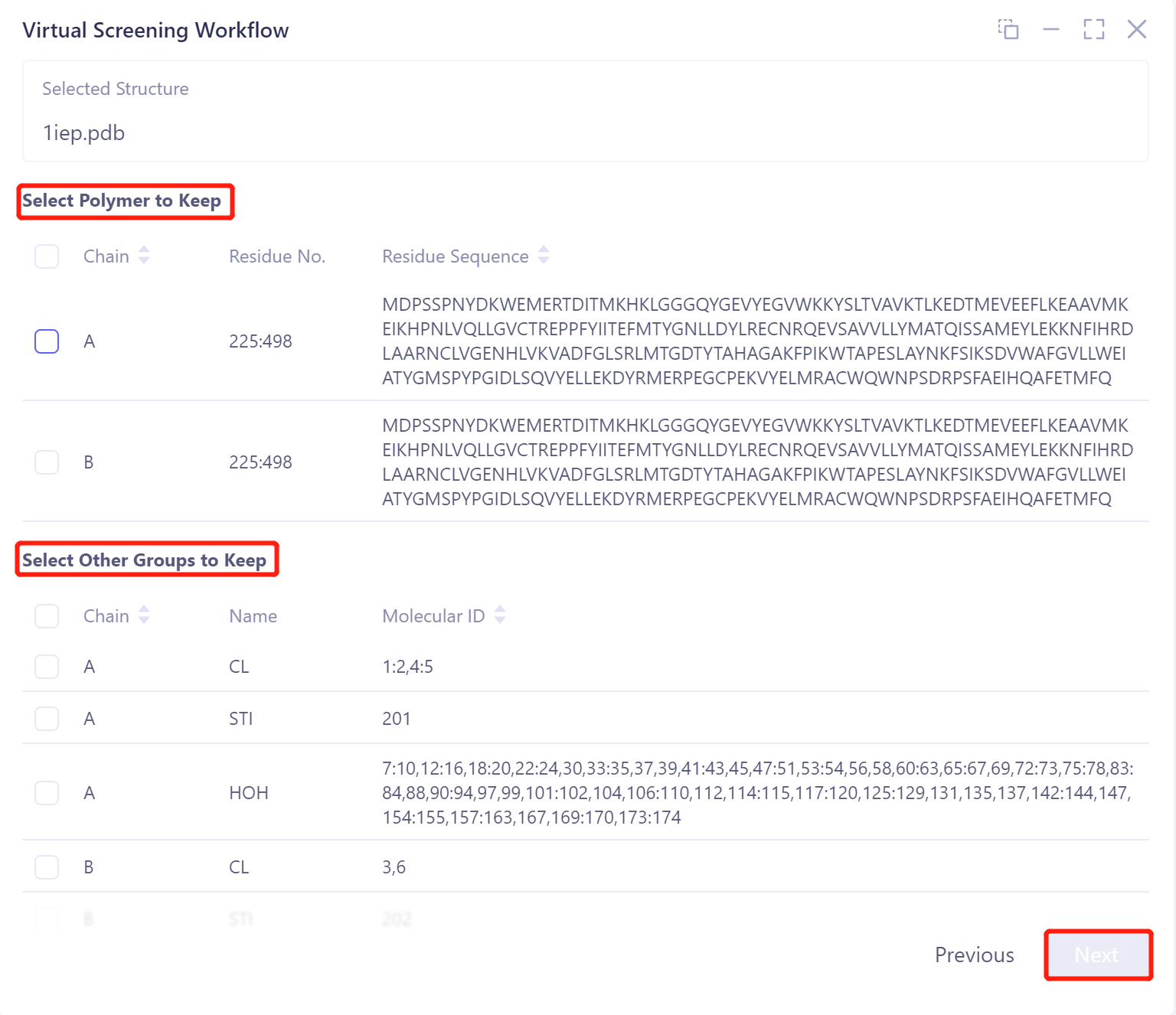
Select Polymer to Keep:选择需要保留的链。
-
Select Other Groups to Keep:选择需要保留的其他基团。
-
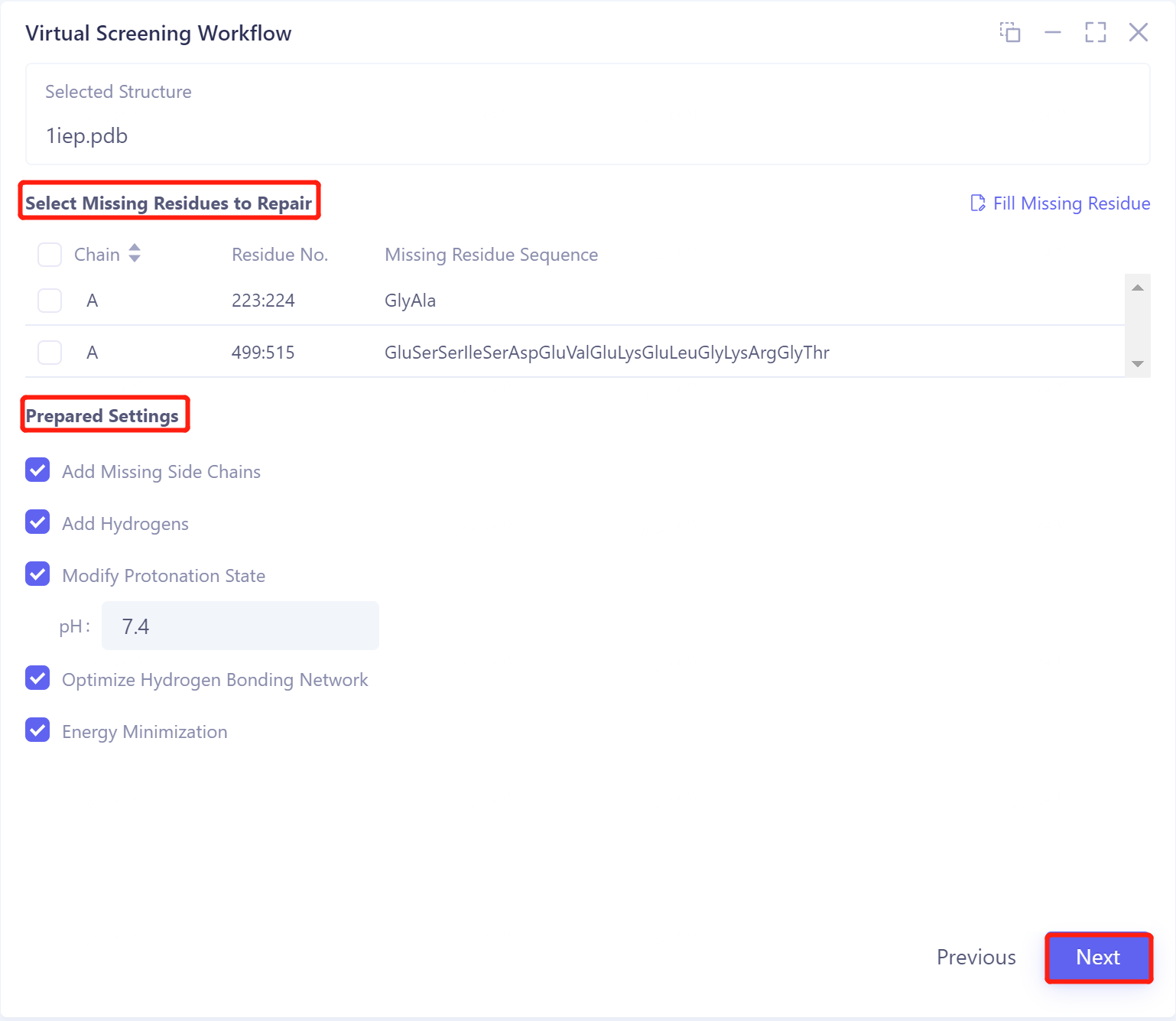
Select Missing Residues to Repair:
-
可以根据.pdb文件内的序列相关数据来进行修复;
-
如点击“Fill Missing Residue”,可以选择上传.fasta文件对缺失的信息进行修复。
-
-
Prepared Settings
-
Add Missing Side Chains:添加缺失侧链;
-
Add Hydrogens:加氢;
-
Modify Protonation State:调节蛋白质环境pH,使蛋白质达到该pH下的质子化状态;
-
Optimize Hydrogen Bonding Network:优化氢键网络;
-
Energy Minimization:能量最小化。
-
2.2.2 配体准备
-
配体文件输入,共有四种方式:
-
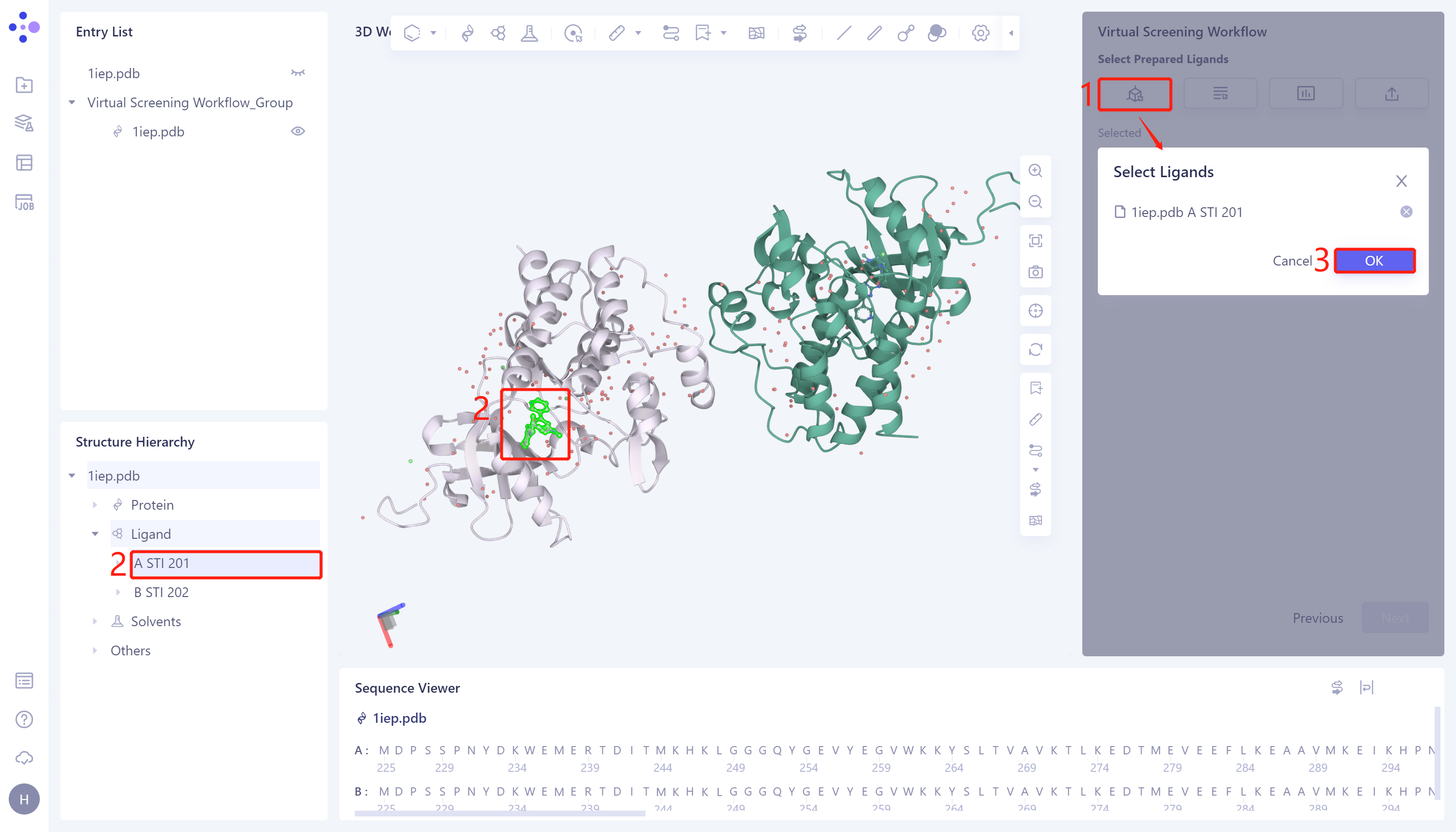
1)Select from 3D:点击Select from 3D 选框 → 弹出Select Ligands框 → 界面左侧Structure Hierarchy/3D Workspace窗口内选择所需配体结构 → Select Structure框内显示选中的配体名称,点击OK。
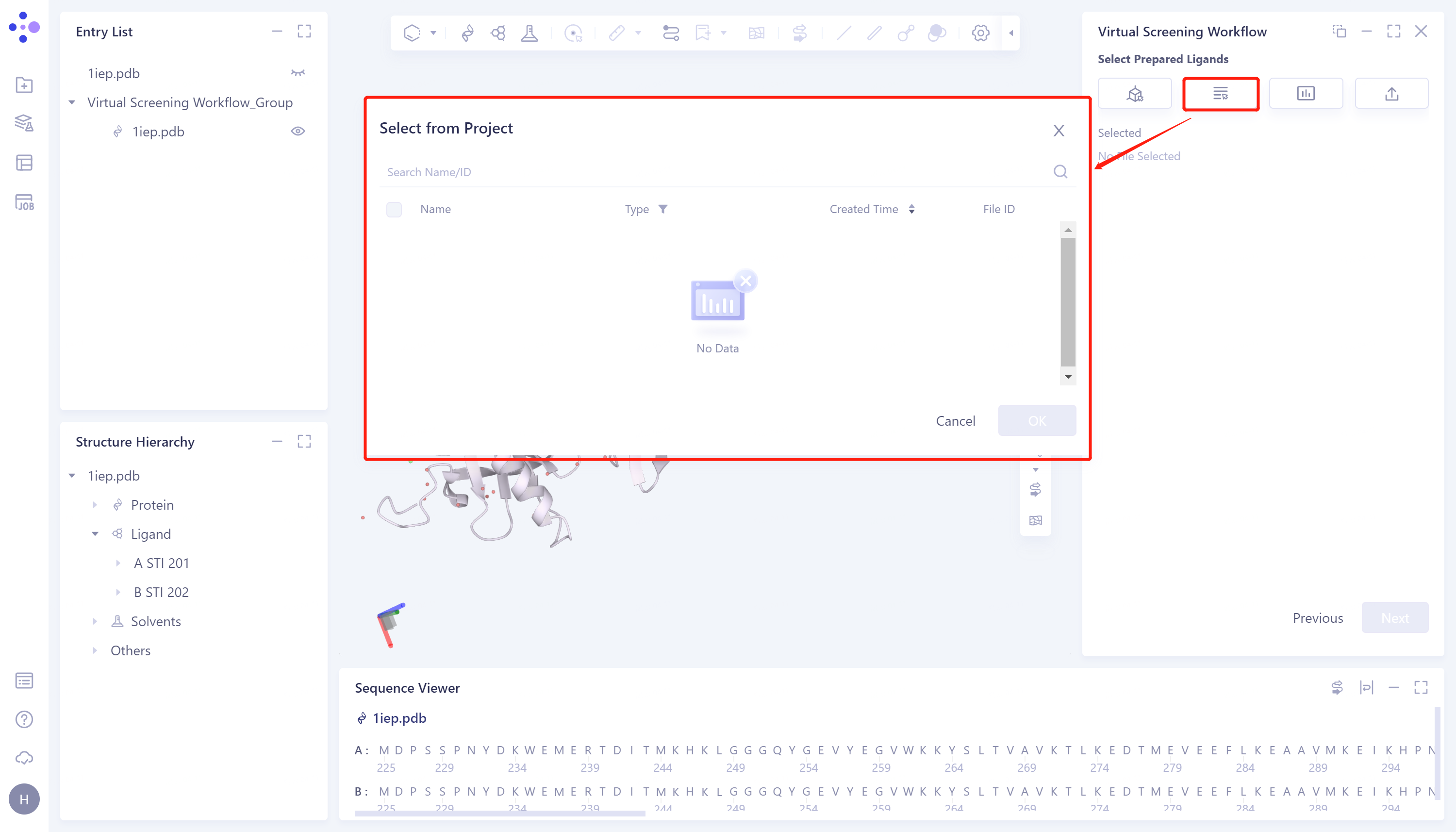
- 2)Select from Project:点击Select from Project选框 → 界面中间弹出Select from Project界面 → 选中所需配体结构 → 点击OK。
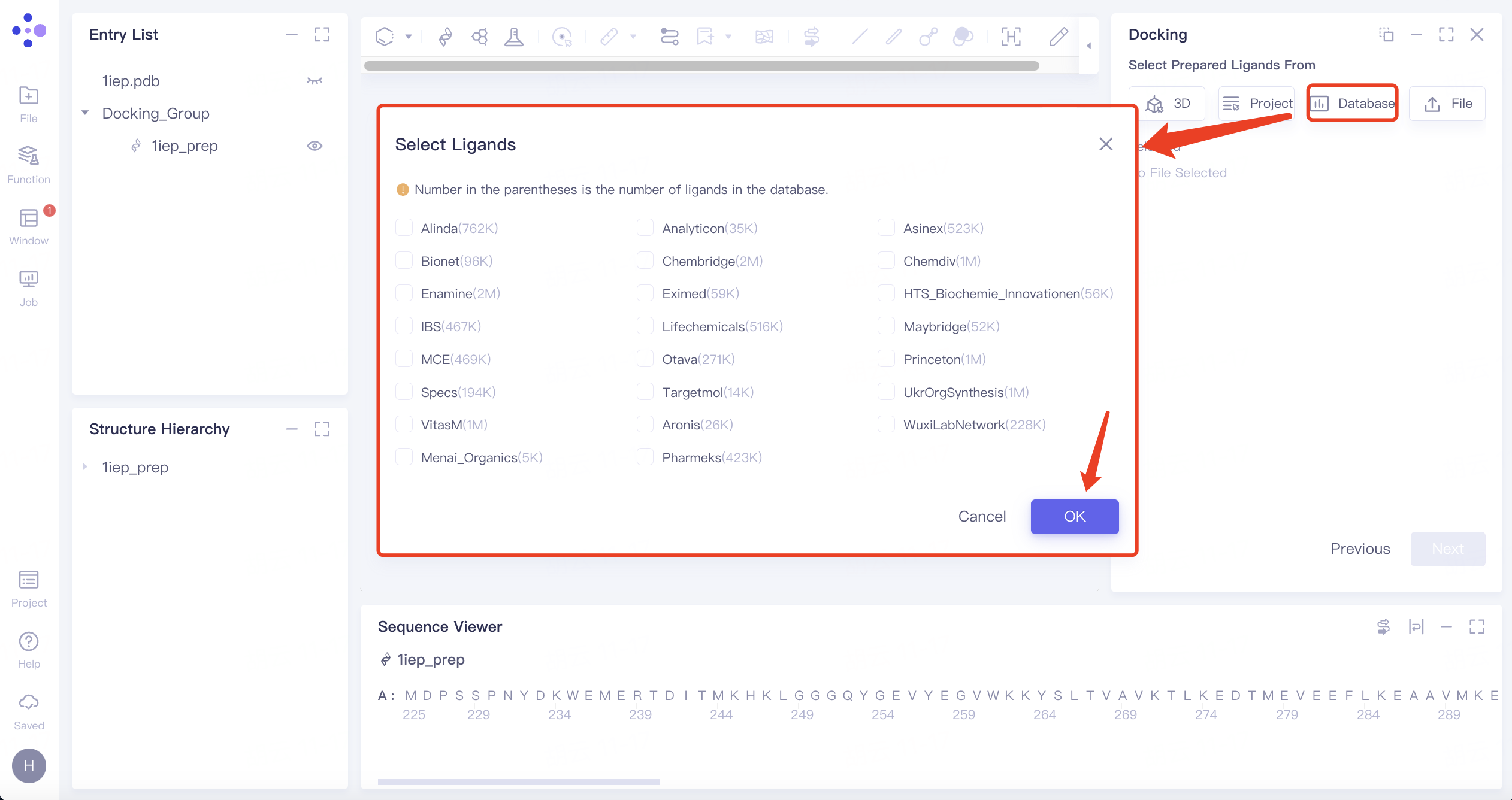
- 3)Select from Database:点击Select from Database选框 → 界面中间弹出Select Ligands界面 → 选中所需配体数据集 → 点击OK。
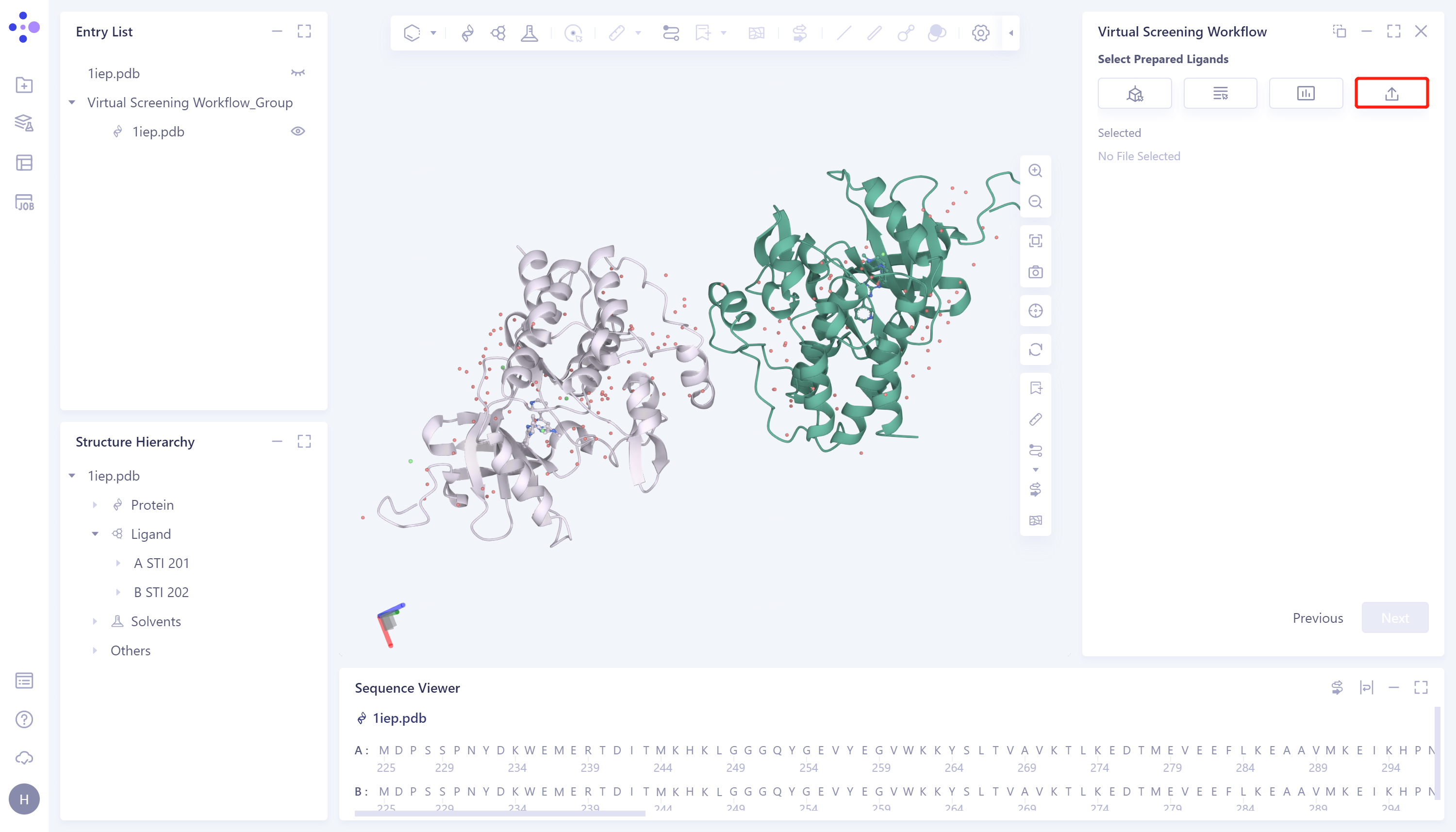
- 4)Select File:从本地文件夹中选择所需配体文件(支持.mol和.sdf文件格式)并上传。
-
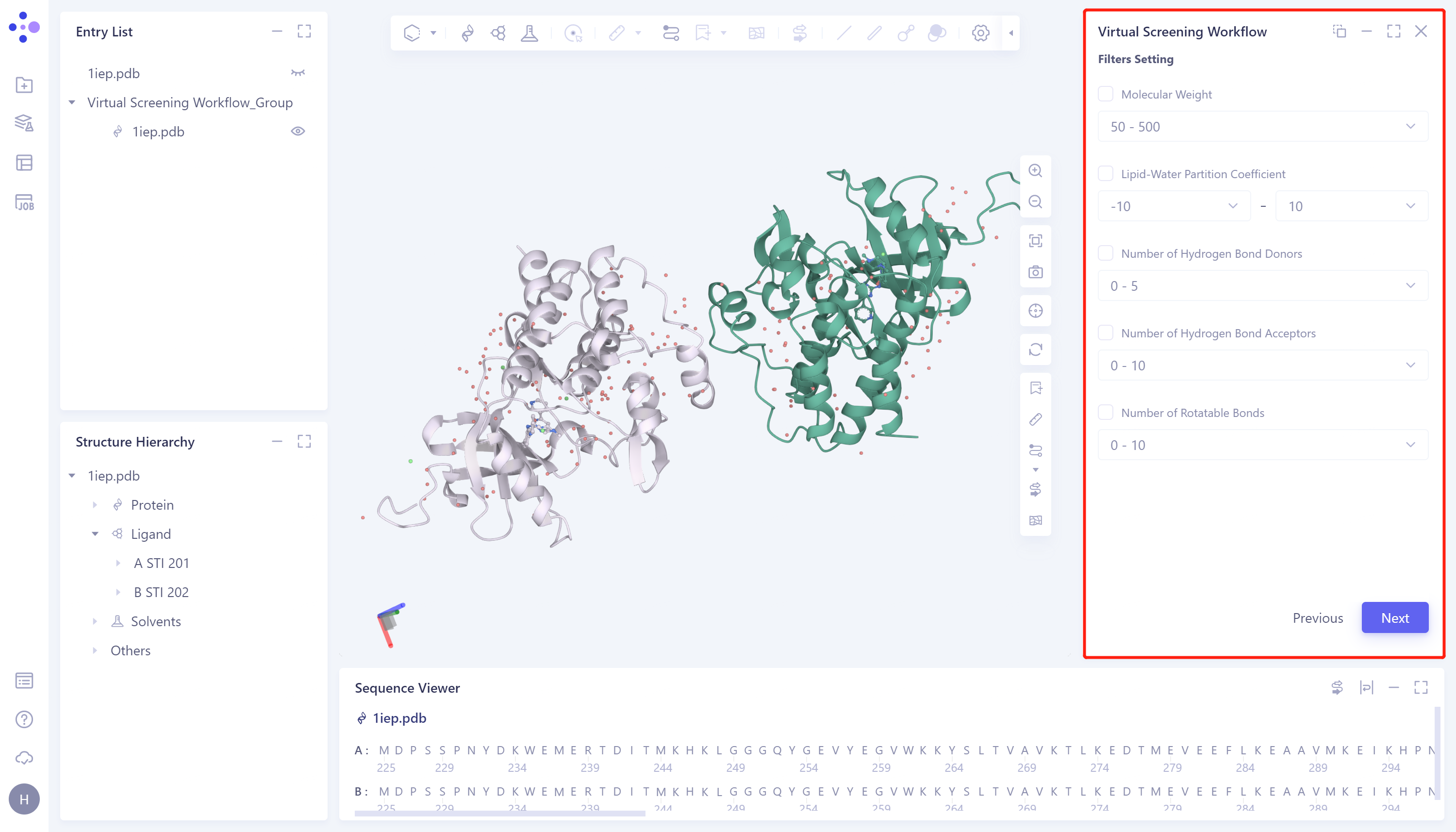
设置筛选条件:如果您对所筛选分子的性质有限制要求,可通过Filter Setting窗口提供的性质约束筛选的分子范围。具体操作为:勾选所需限定范围的分子性质 → 调整范围 → Next进入下一步。如果无特殊要求,则直接点击Next跳过该步骤。
-
Molecular Weight:分子质量;
-
Lipid-Water Partition Coefficient:脂水分配系数;
-
Number of Hydrogen Bond Donors:氢键供体数;
-
Number of Hydrogen Bond Acceptors:氢键受体数;
-
Number of Rotatable Bonds:可旋转键数。
-
2.2.3 对接盒子设置
-
定义对接盒子有5种方式,如下:
-
1)Select Ligand in the structure as center:该方法以3D Workspace窗口显示的配体的所在区域定义对接盒子。具体步骤为:点击Select Ligand in the structure as center选框 → Select Ligand Name中选择配体名称(仅当3D Workspace窗口显示配体时,Select Ligand Name中才会给出配体选项),选中后则自动生成盒子 → Cutoff设置盒子大小,值越大盒子越大,设置完成后点击Apply应用至当前盒子 → Center处可手动设置盒子中心的坐标 → Size处可手动设置盒子大小 → 点击Next进入下一步。

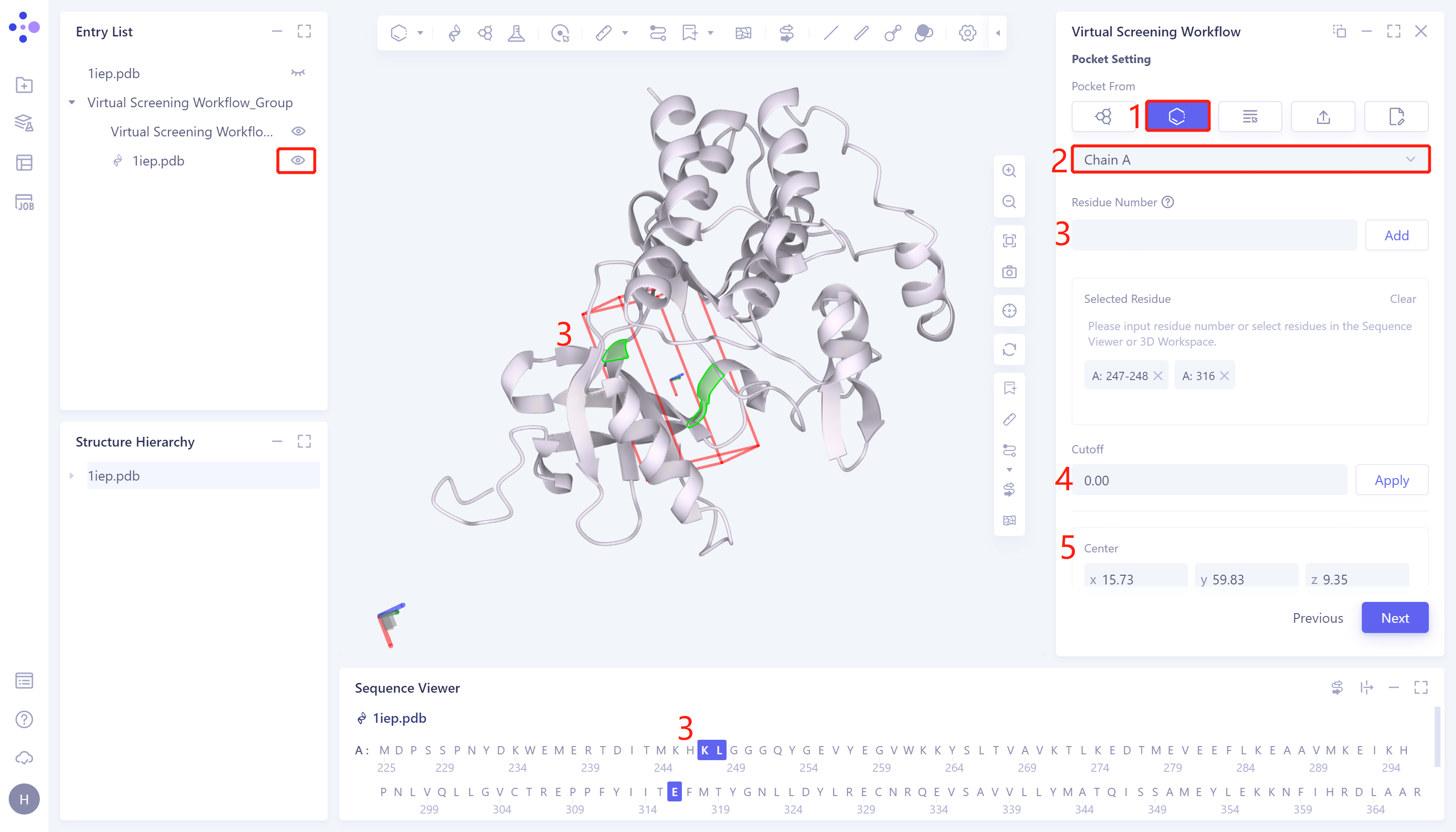
- 2)Select Residues as Center:该方法通过选择蛋白质序列中的氨基酸残基生成对接盒子。具体步骤为:点击Select Residues as Center选框 → Select Residue Chain ID选框中选择蛋白链名称(仅当该任务下的蛋白受体显示于3D Workspace窗口时,Select Residue Chain ID选框中才会给出选项) → 通过Residue Number(输入残基对应的序号,点击Add)/3D Workspace窗口(点击对接盒子边缘的残基从而指定盒子位置)/Sequence Viewer窗口(点击残基)选择氨基酸残基,选中的氨基酸残基自动显示于Selected Residue的框中,并自动生成对接盒子 → Cutoff设置盒子大小,值越大盒子越大,设置完成后点击Apply应用至当前盒子 → Center处可手动调整盒子中心的坐标 → Size处可手动设置盒子大小 → 点击Next进入下一步。
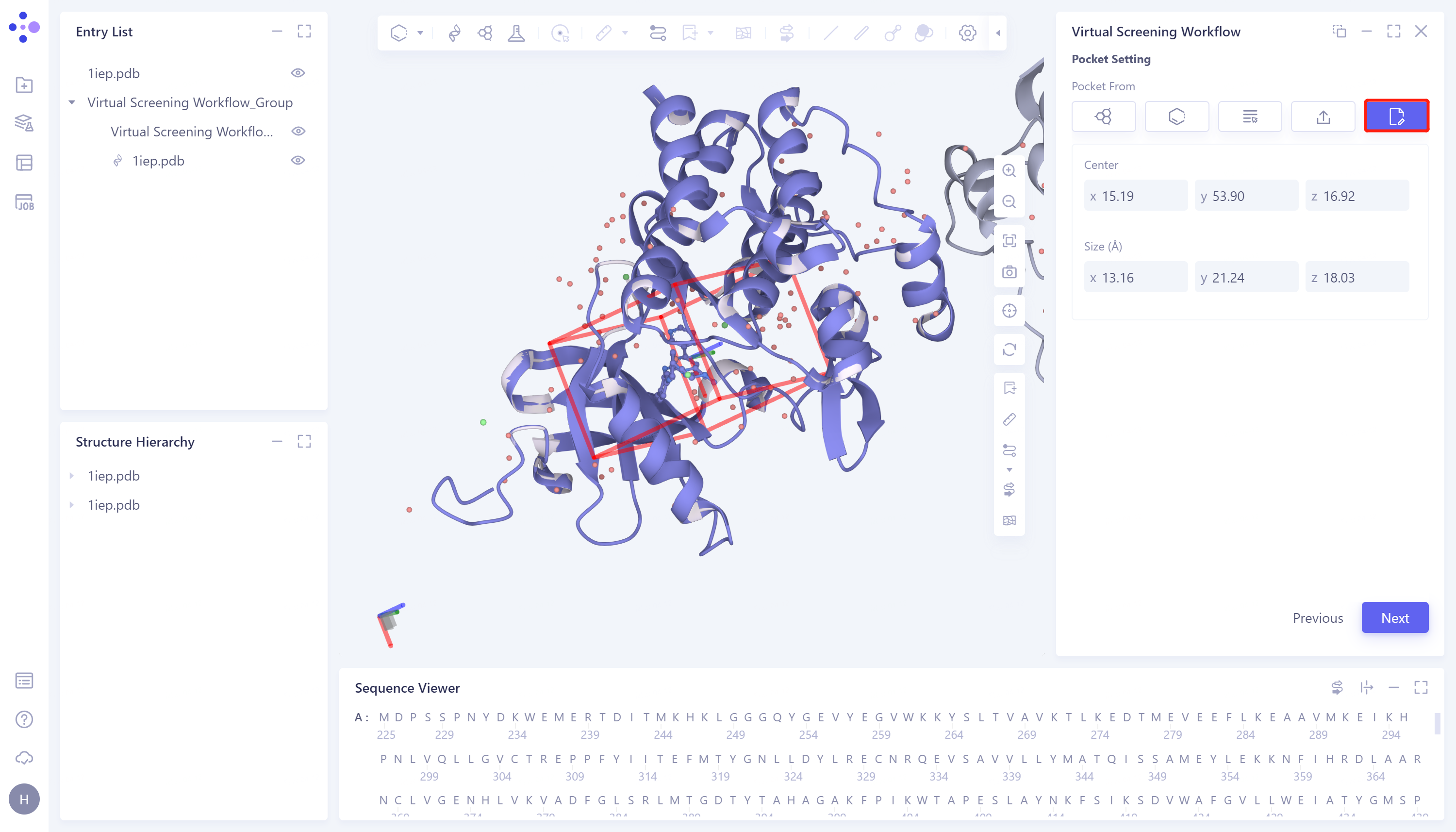
- 3)Select Pocket from Project:该方法选择Hermite平台的项目中的盒子作为本次任务中的对接盒子。具体步骤为:点击Select Pocket from Project选框 → 弹出的Select Pocket from Project框中选择所需盒子 → 点击OK → Center处可手动调整盒子中心的坐标 → Size处可手动设置盒子大小 → 点击Next进入下一步。

- 4)Select File:支持导入本地文件中的盒子文件(.txt格式)。具体步骤为:点击Select File选框 → 选择本地文件中的盒子文件(.txt文件) → Center处可手动调整盒子中心的坐标 → Size处可手动设置盒子大小 → 点击Next进入下一步。
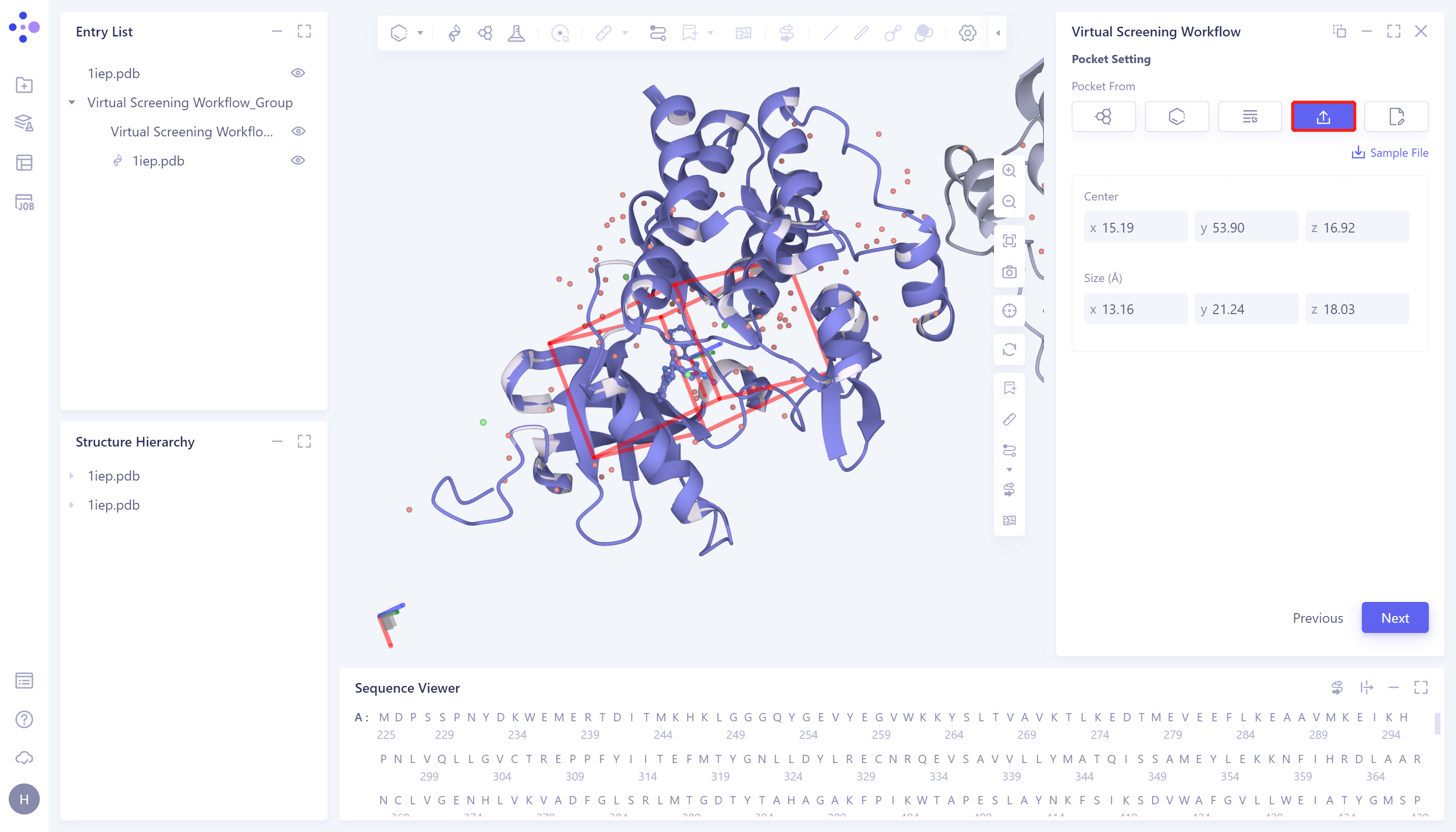
- 5)Input:直接手动输入盒子参数。具体步骤为:点击Input选框 → Center处设置盒子中心的坐标 → Size处设置盒子大小 → 点击Next进入下一步。
- 注:盒子体积不得小于1000ų。
2.2.4 对接参数
-
搜索模式,可选Fast Mode Docking、Balanced Mode Docking、Detailed Mode Docking,可调参数:
-
Number of Results to Keep to 1st/2nd/3rd Docking,可选All/Top Number/Top Percent。
-
Keep Multi Binding Poses for Each Ligand:每个配体生成多个构象
-
Number of Binding Pose:生成的构象个数,支持数量1~10;
-
Energy Range (kcal/mol):与最优结合模型相差的最大能量值,支持1~9;
-
Do MM PB/GBSA Structure:MM PB/GBSA Structure最多能处理3000个配体。
-
打分函数(Scoring Function),可选Vina、Vinardo、AutoDock4。
-

-
如果前一步骤勾选了Do MM PB/GBSA Structure,则进入该界面设置MM PB/GBSA的参数:
-
Solvation Mode:确定表面积生成方式,支持PB和GB模型 可选PBSA或者GBSA;
-
确定蛋白和配体力场,设置计算参数:
- 红框:选择蛋白力场,可选amber03、amber99sb、amber99sb-ildn、amber99sb-star-ildn-mut;
绿框:选择配体力场,可选gaff2、gaff;
黄框:选择是否能量最小化;
蓝框:配置溶剂环境参数(Dielectric Constant、Implicit Solvent Dielectric Constant、Non-polar Surface Constant、Non-polar Surface Coefficient)。 - (注意:强烈建议保留默认参数)
- 红框:选择蛋白力场,可选amber03、amber99sb、amber99sb-ildn、amber99sb-star-ildn-mut;
-
Total Decomposition Contribution Analysis:计算氨基酸残基的能量贡献。
-

2.2.5 确认
-
确认输入的蛋白及配体文件;
-
Pocket Config处确认对接盒子位置及大小;
-
Scoring Function处确认选用的打分函数;
-
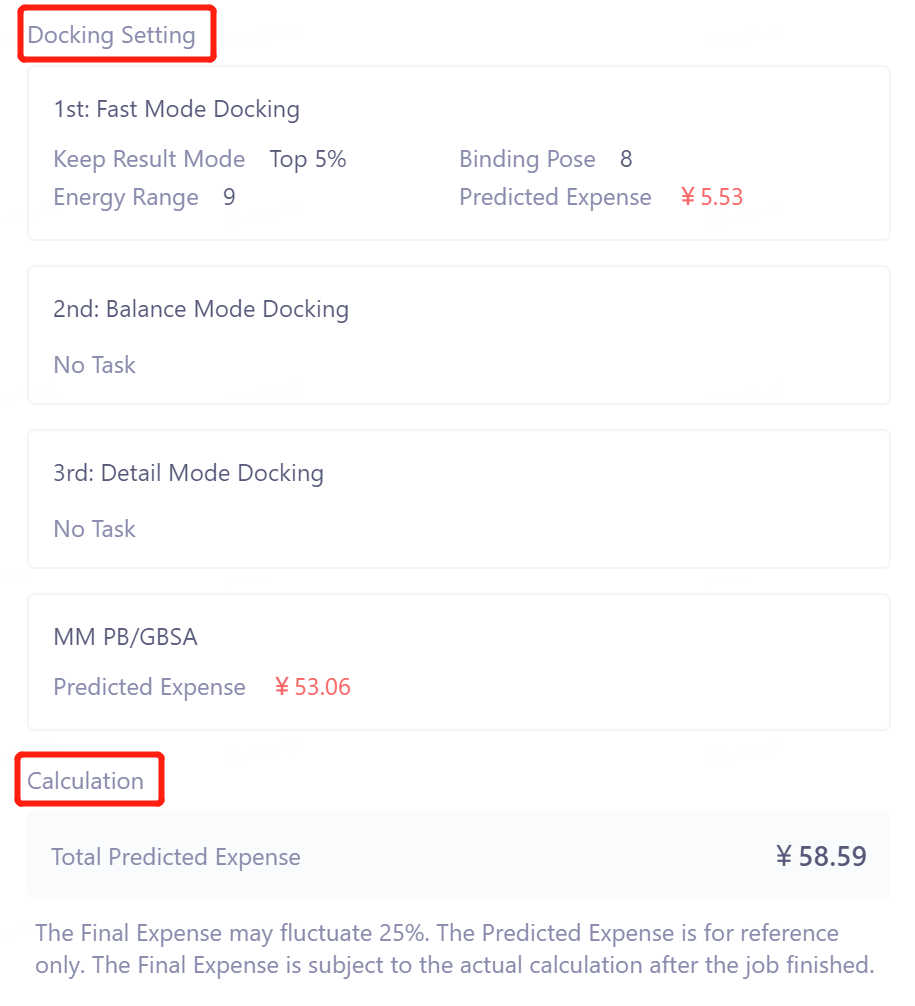
Docking Setting处确认对接参数及计算花费;
-
Calculation处的Total Predicted Expense给出了该任务的总花费。

-
Job Name处命名该任务;
-
点击Submit提交任务。
3. 结果展示
3.1 入口
-
左侧通用菜单栏Menu Job → 找到所需任务。
-
可以通过搜索Job Name找到该任务,也可以通过Job Type的筛选找到。
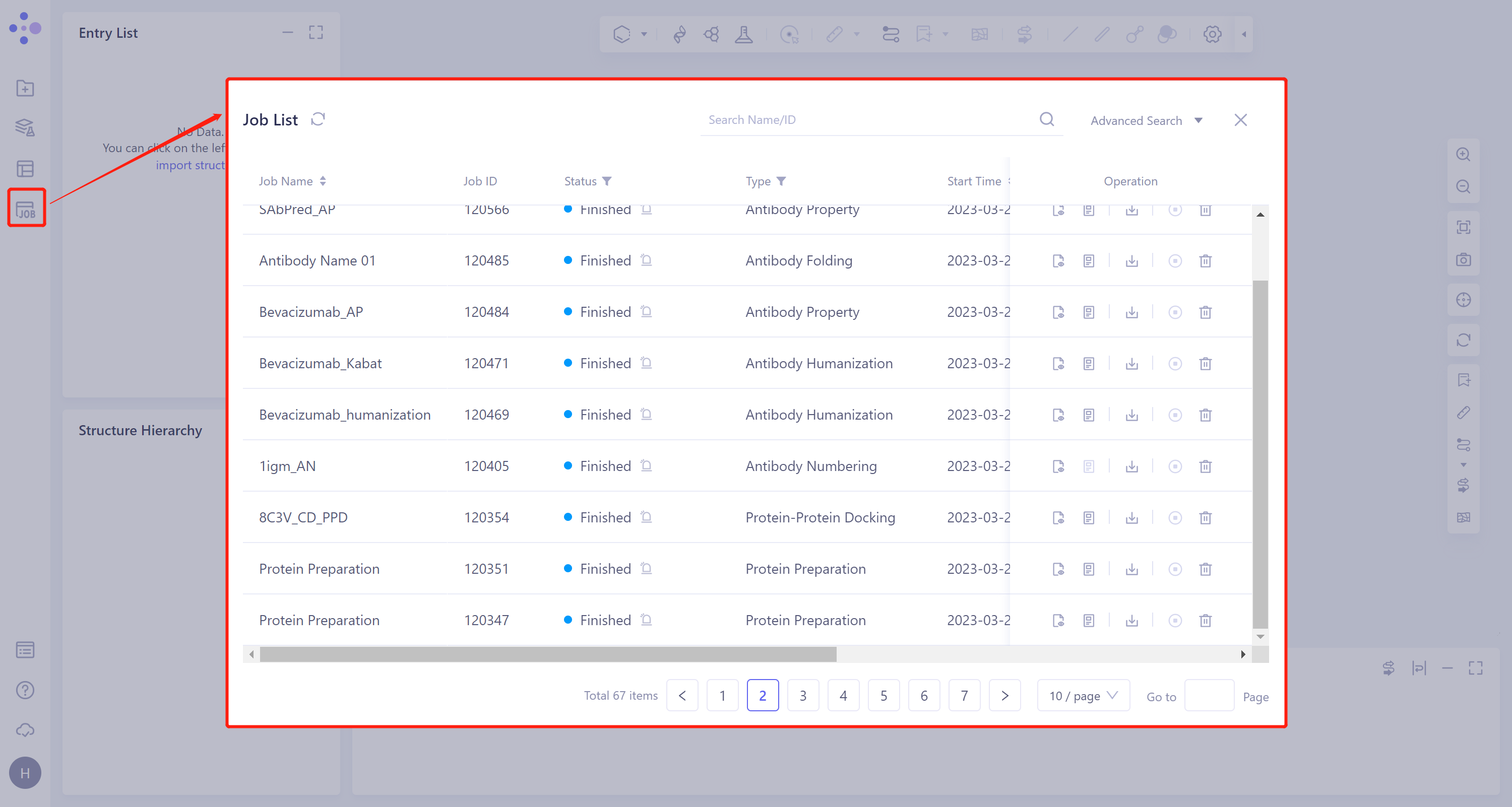
- 该任务产生四类子任务:Protein Preparation、Ligand Preparation(当输入配体为平台提供的Database时则不产生该任务)、Docking(包含Fast、Balance和Detail,与提交任务的勾选情况有关)和MM PB/GBSA Structure(勾选计算后产生),分别记录着蛋白准备的结果、虚拟筛选的结果及MM PB/GBSA结果。
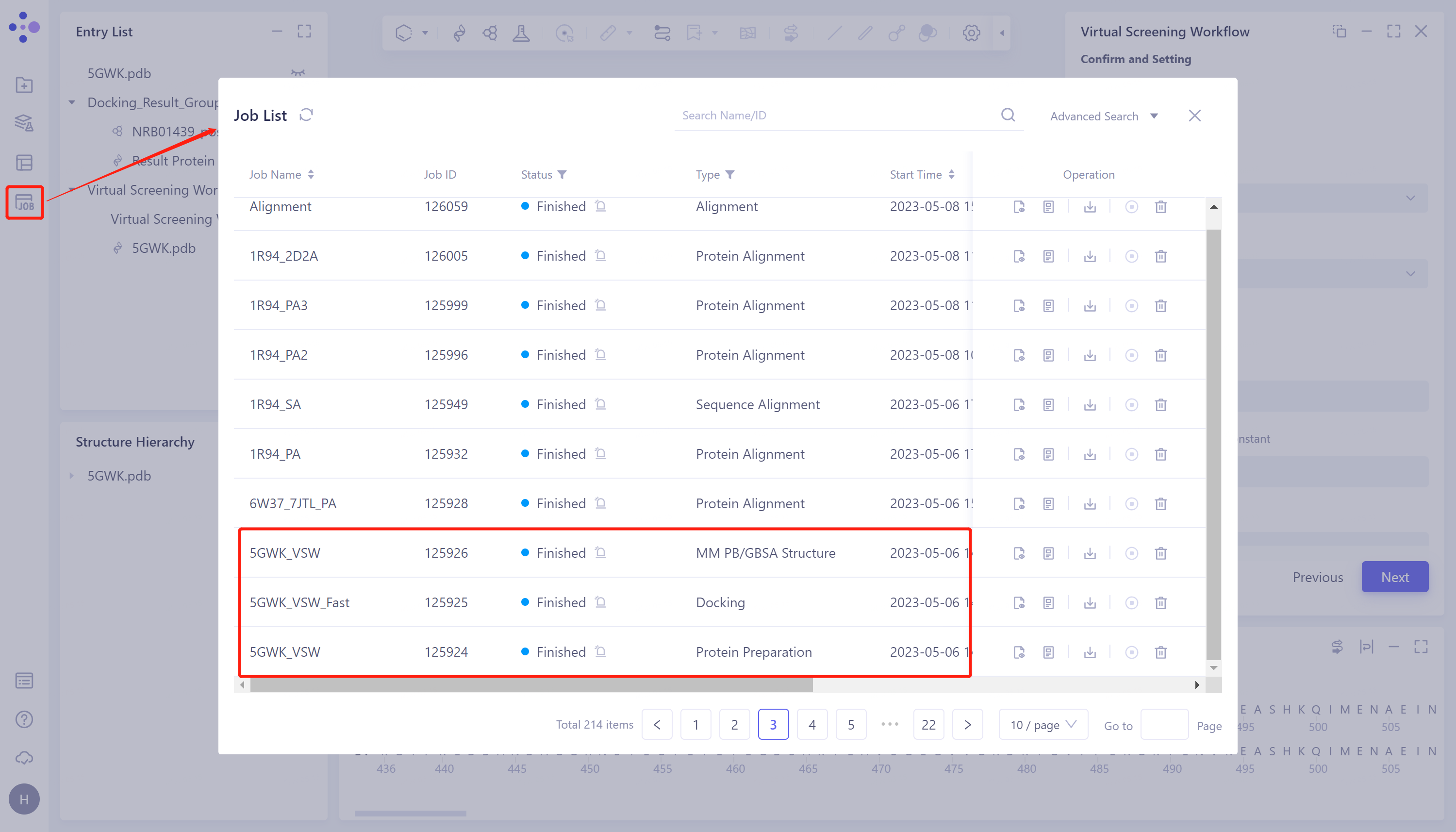
3.2 Docking结果展示
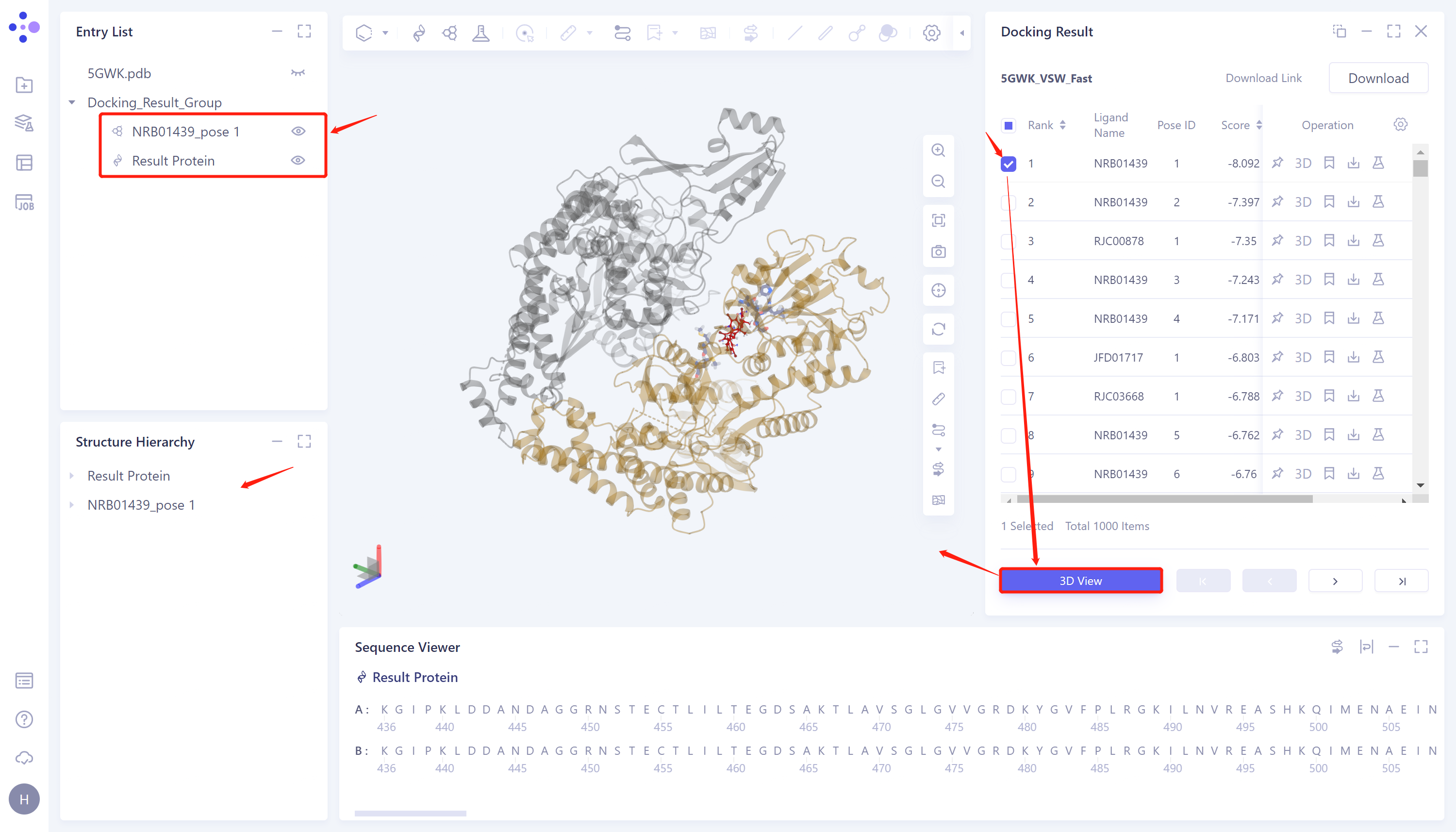
- 选择需要查看的任务,点击Operation列中的Show显示该任务的结果,界面如图所示。
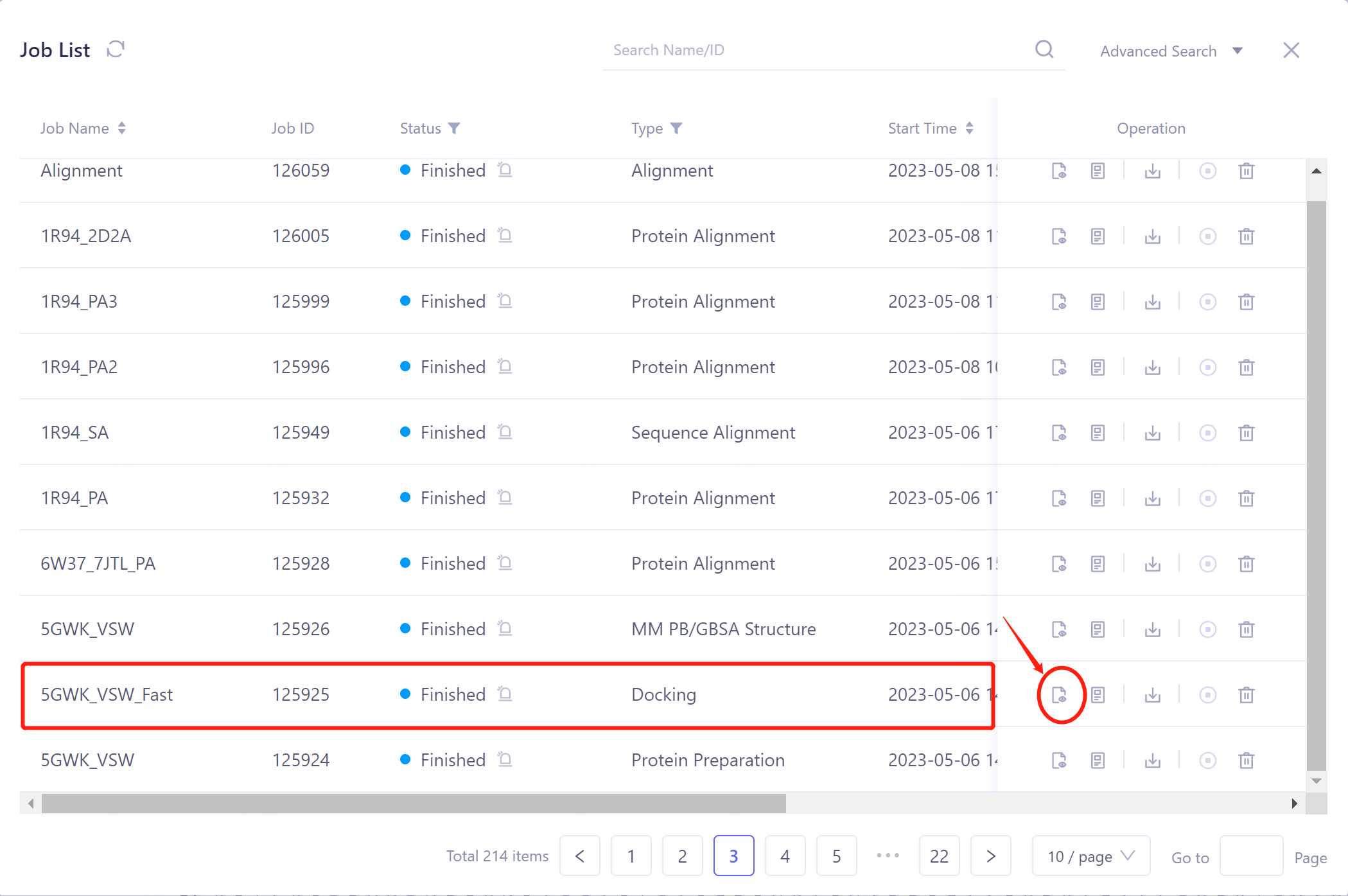
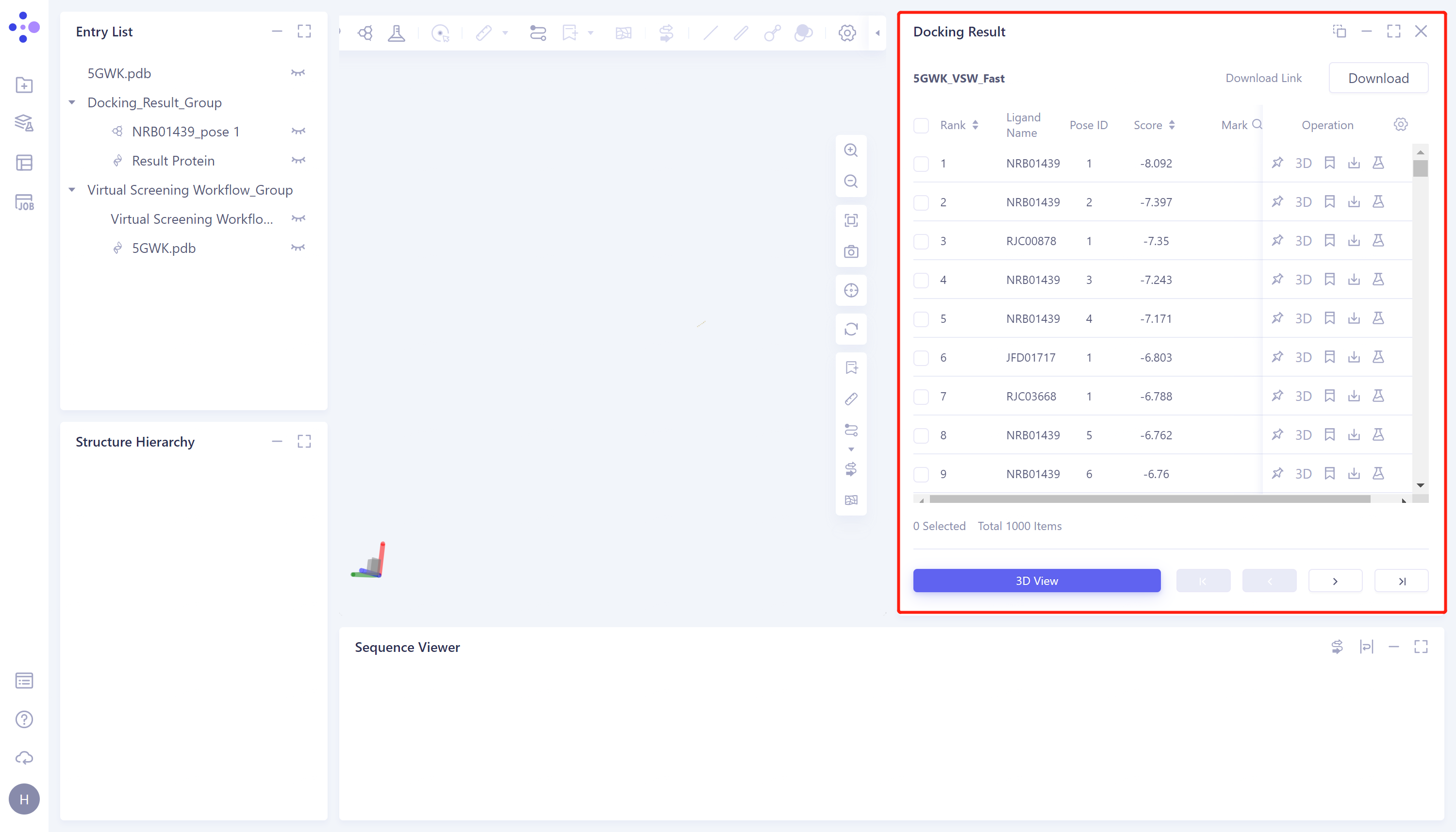
-
Docking Result表格说明:
-
Ligand Name记录了配体名称;
-
Pose ID记录配体结合模式的序号;
-
Score为配体与蛋白的对接评分,点击即可按从高到低或者从低到高的方式进行排序;
-
Operation下有5个操作选项,可对该行分子进行操作:
-
Fix:在3D Workspace中固定显示。
-
3D:在3D Workspace中显示该配体。
-
Mark:针对部分特殊的数据行,可以点击Mark处进行人工备注,备注好后会显示于Mark一栏中,并支持按备注搜索。
-
Download:下载对接后的该分子,可储存为sdf、mol和mol2格式。
-
Properties:Ligand自带的非规范Property。
-
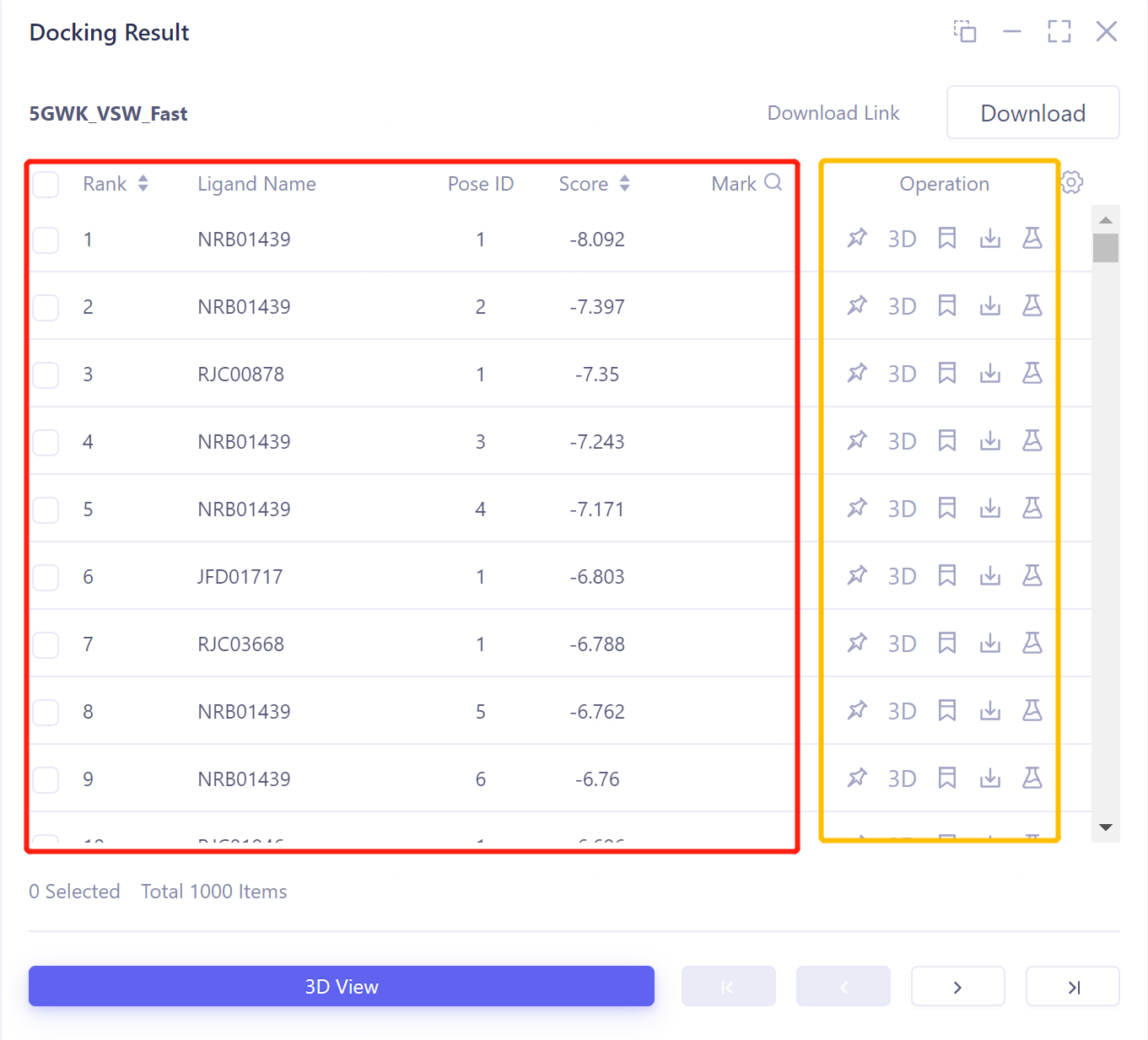
-
图像说明:
-
3D View:将结果显示于3D Workspace窗口中;
-
直接点击3D Viewer将用于对接的蛋白结构显示于3D Workspace窗口中;
-
对接图像显示:右侧Docking Result表格中选中所需展示的配体对应的的复选框 → 点击3D View → 3D Workspace中显示对接结果。
-
-
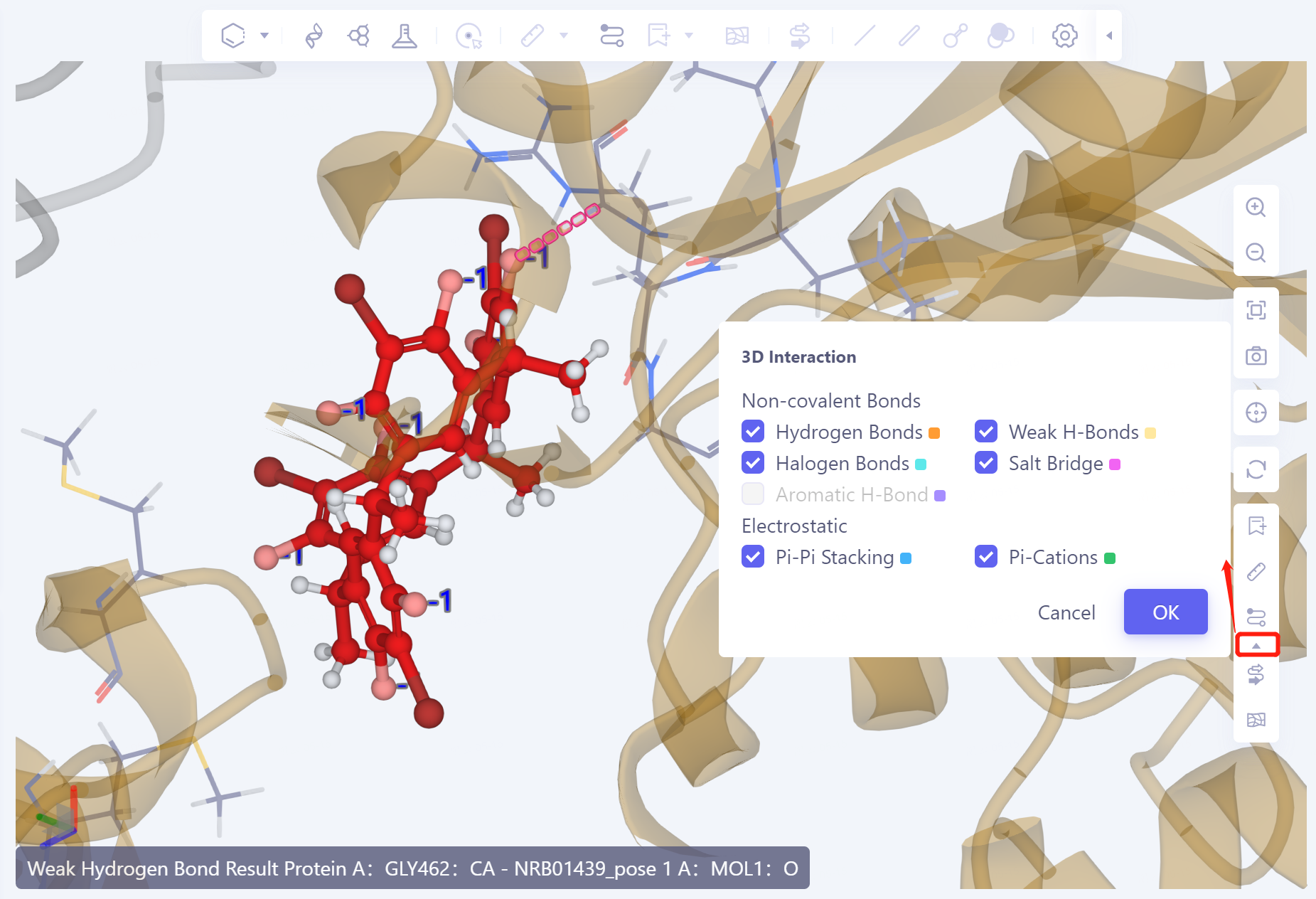
3D Workspace窗口展示:
- 配体以球棍模型显示,与配体具有相互作用的氨基酸残基以Line形式呈现,虚线表示相互作用,点击或悬停至虚线则于3D Workspce窗口左下角显示相互作用说明,不同的相互作用以不同的显色表示。

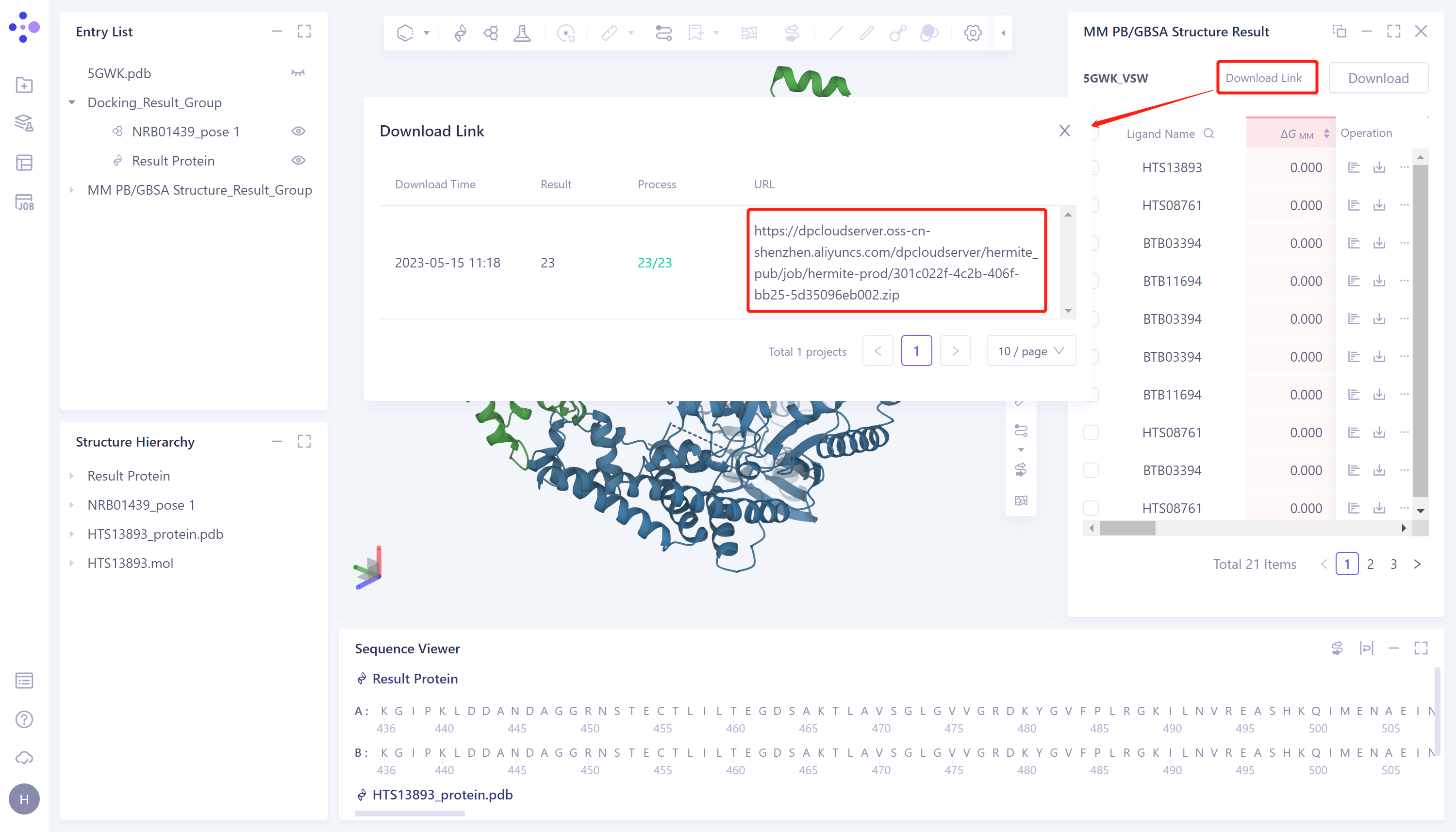
3.3 MM PB/GBSA Structure结果展示
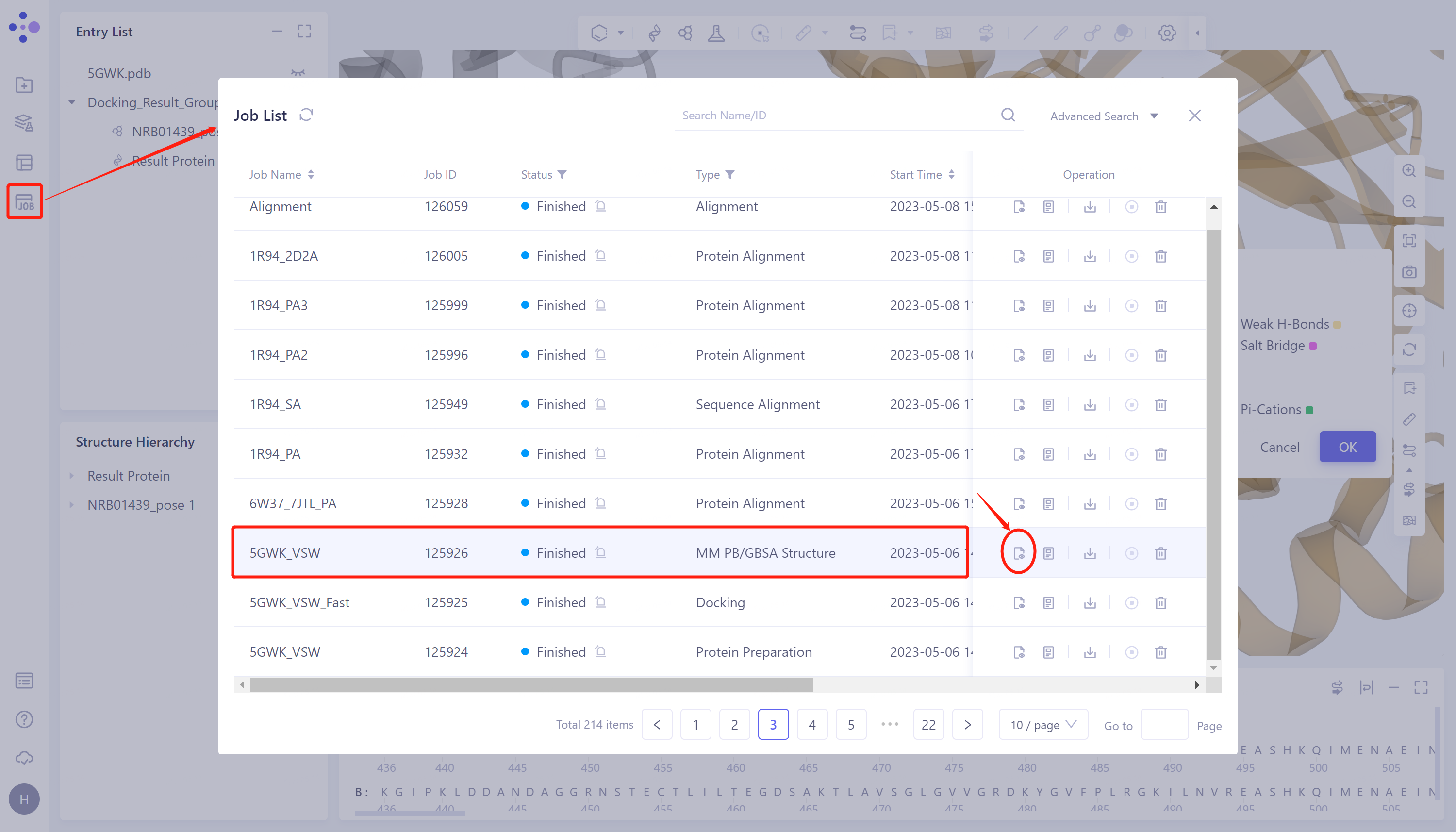
- 左侧通用菜单栏Menu Job → 找到5GWK_VSW的MM PB/GBSA任务 → 点击Operation列中的show展示该任务的结果 → 打开的界面如右图所示。
-
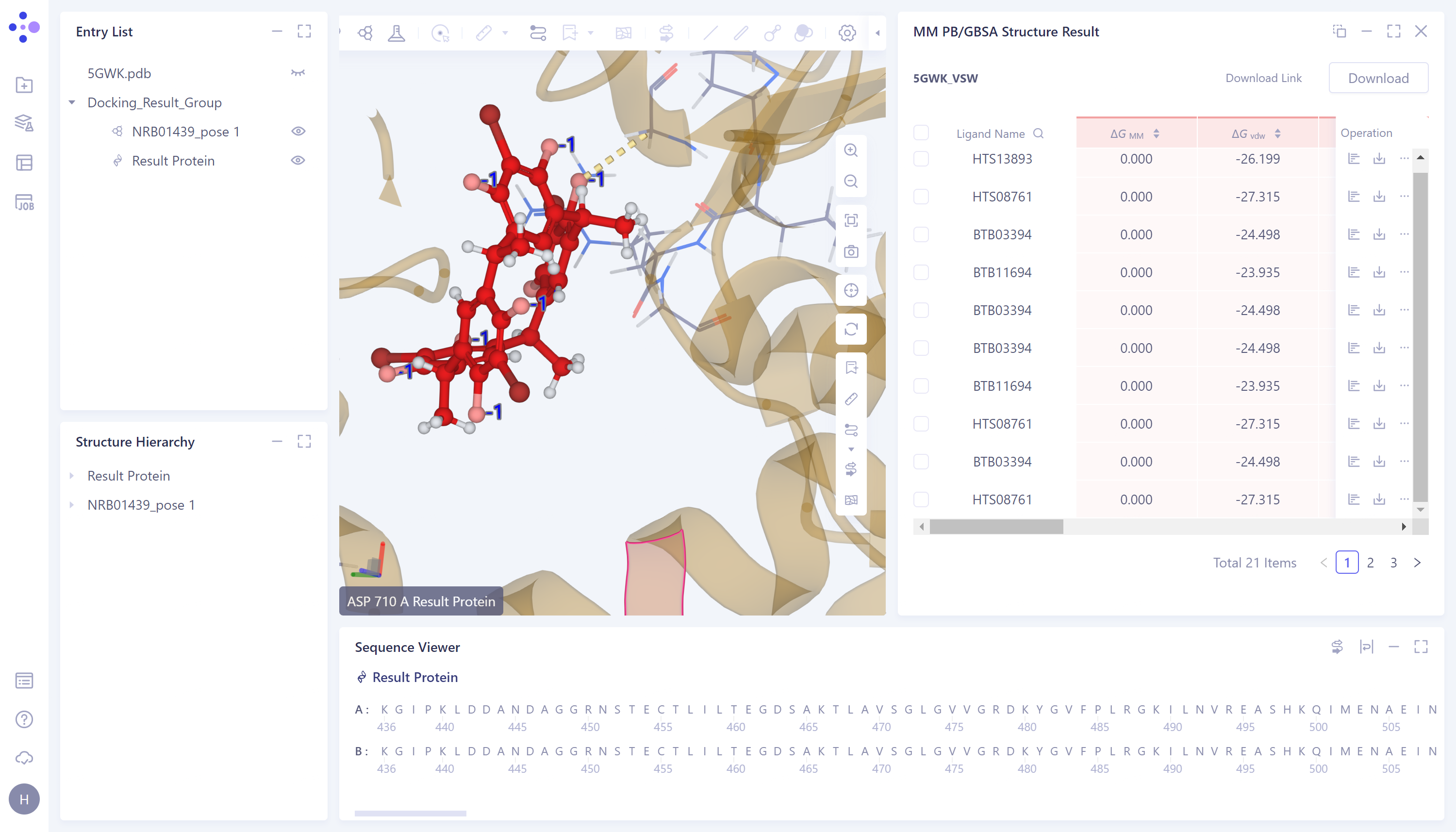
MM PB/GBSA计算结果展示:
-
提供溶剂态、气态和总和3种维度的ΔG数据:
-
-
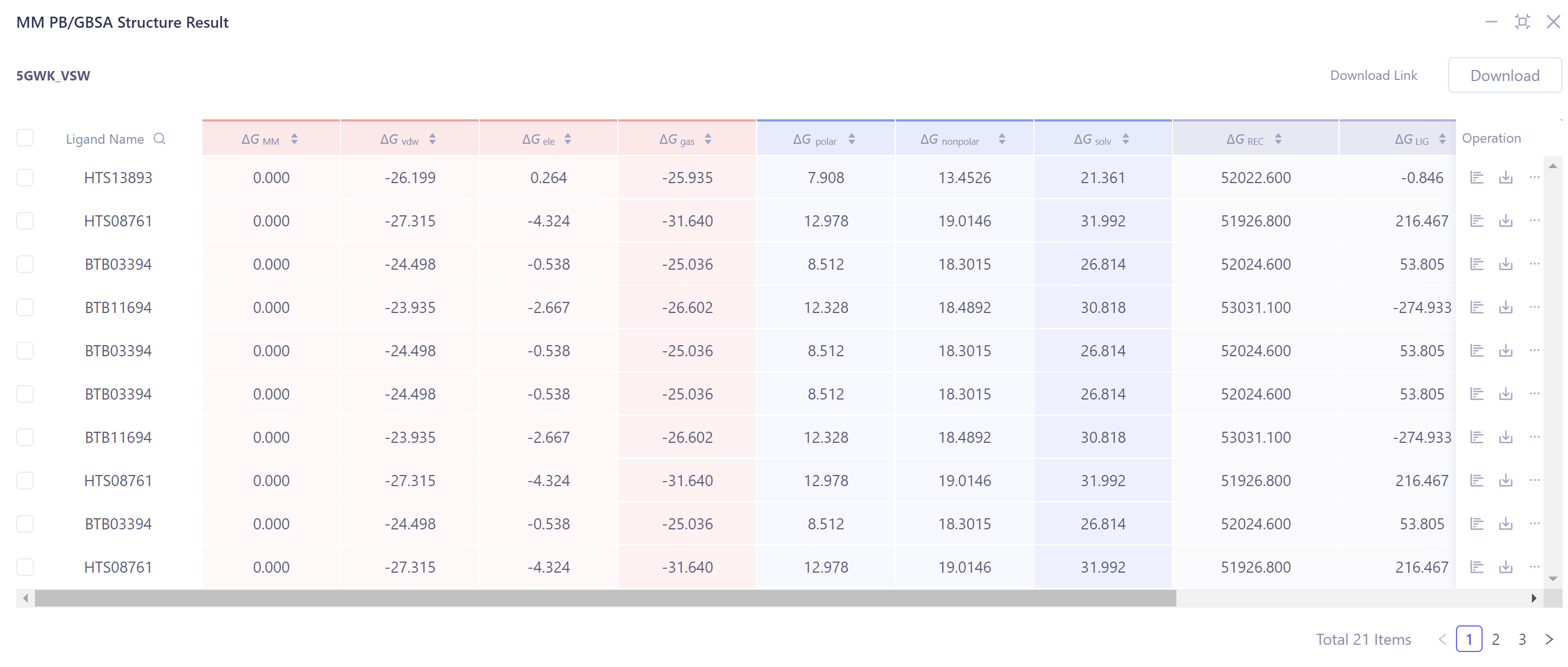
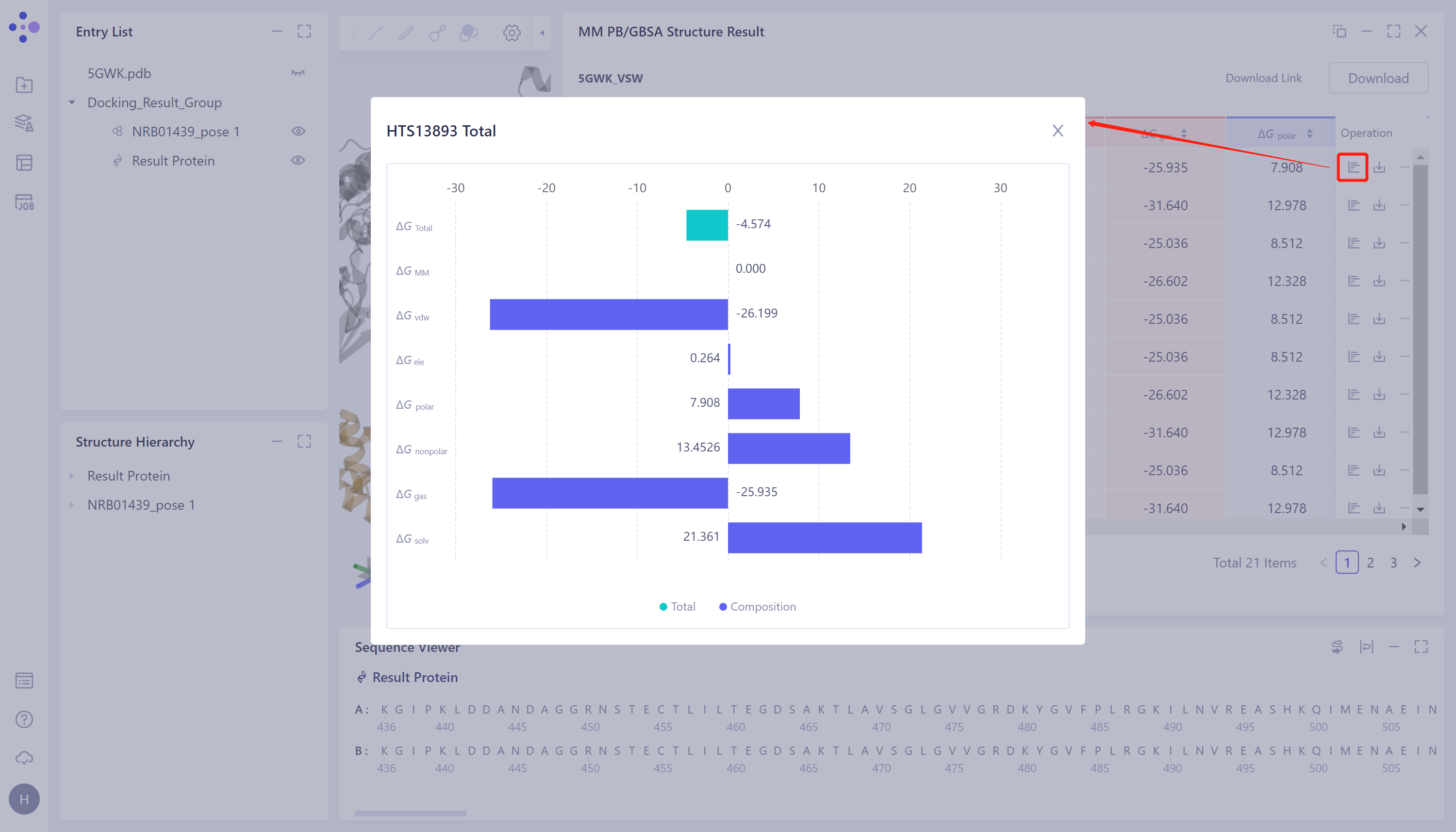
查看ΔG各能量项数值:
- 点击图中红框处按钮,查看和下各个ΔG的分项。
-
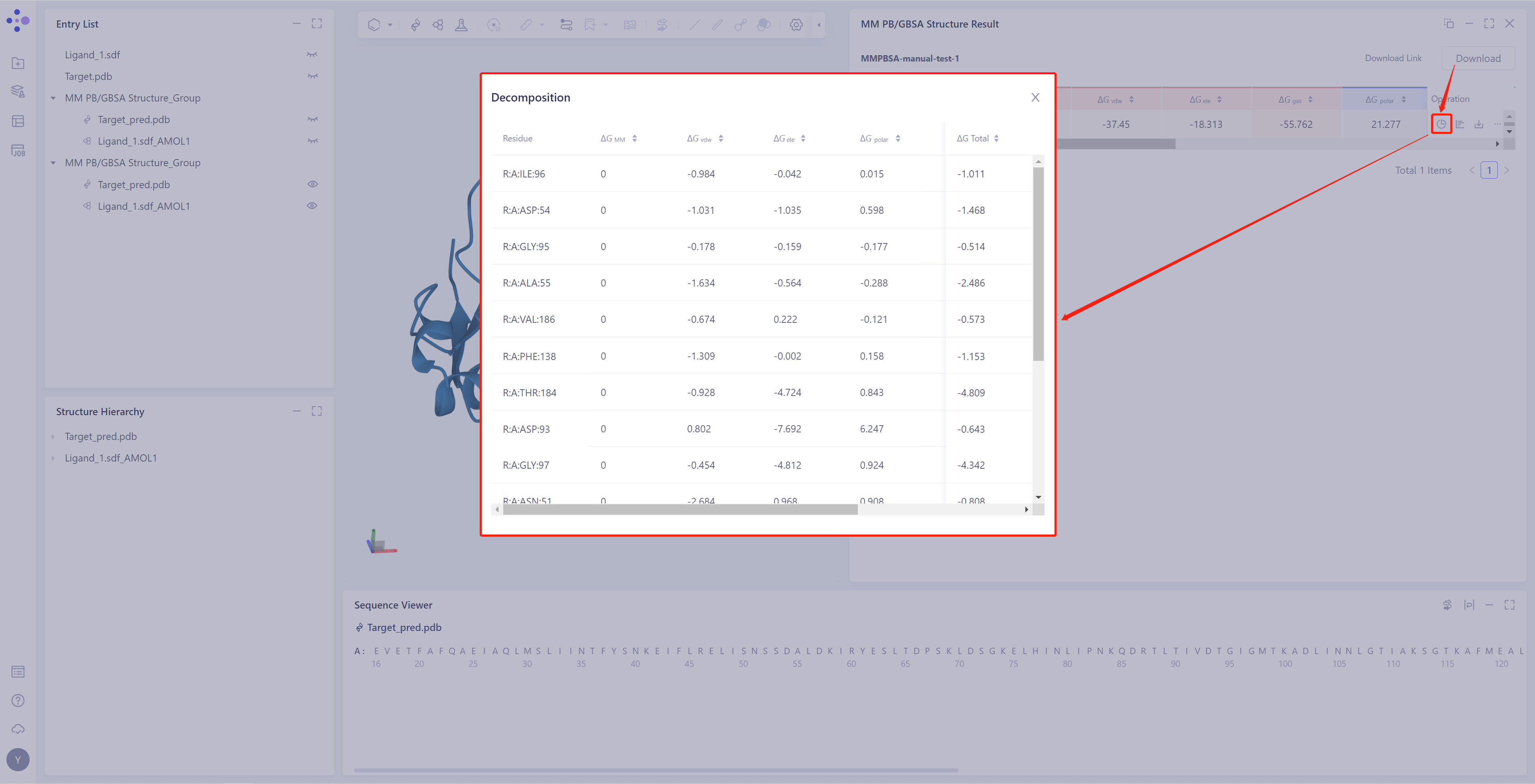
Residue Decomposition:
-
如在计算时,勾选了“Total Decomposition Contribution Analysis”,则可以查看该明细
-
点击图中红框处按钮,按残基进行能量分解,提供残基每一个维度的ΔG数据
-
-
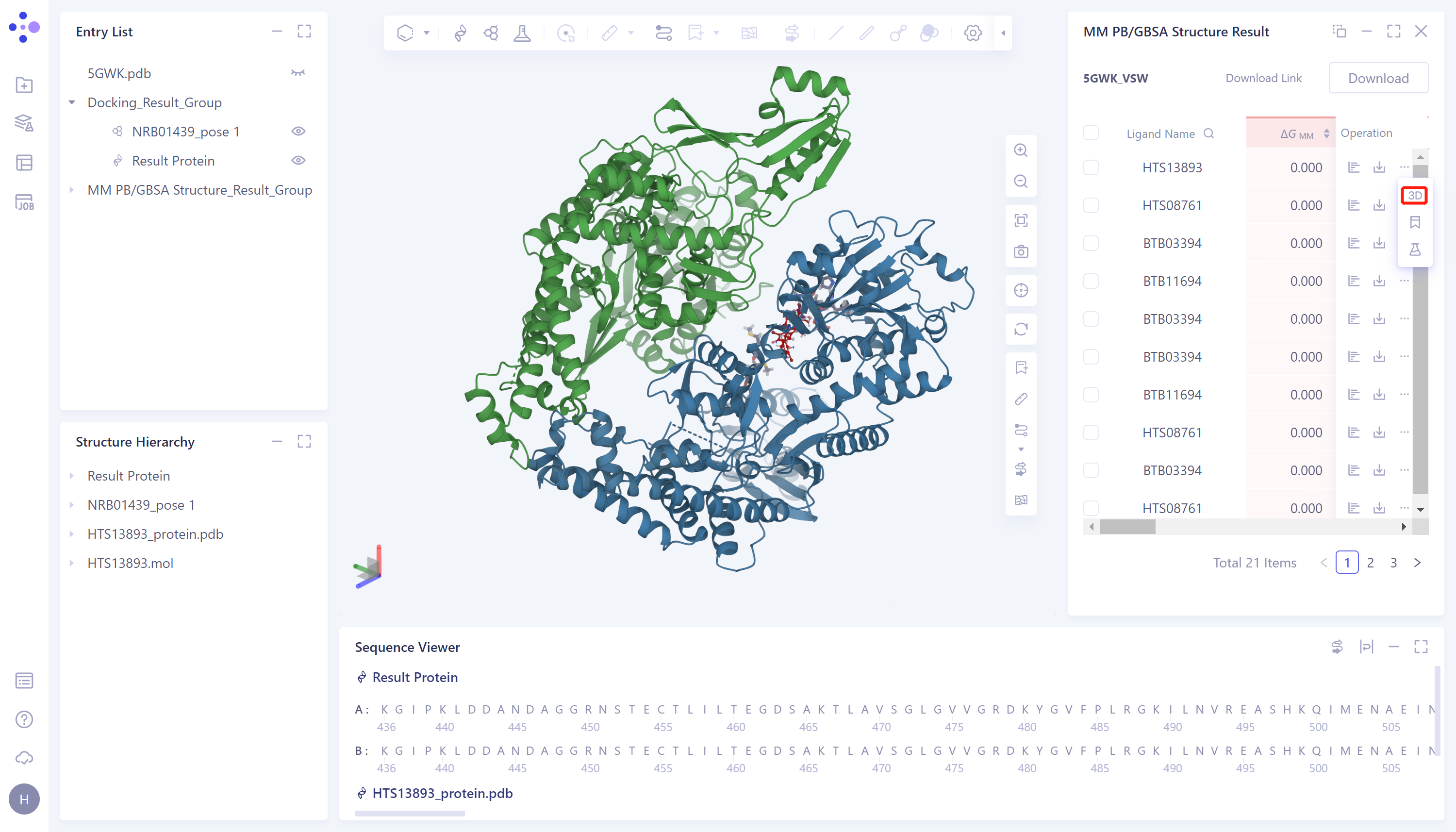
3D Workspace展示:
- 点击图�中红框处“3D”按钮,将对应本行数据的蛋白和配体呈现在3D Workspace中
-
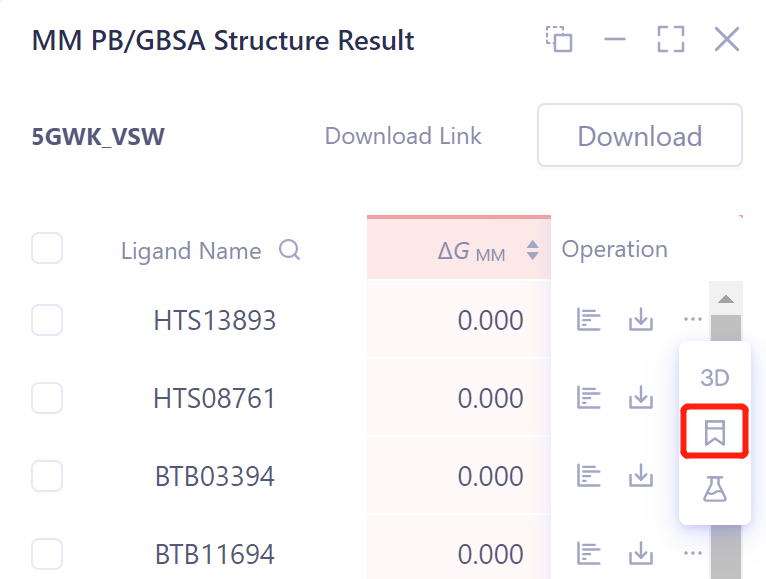
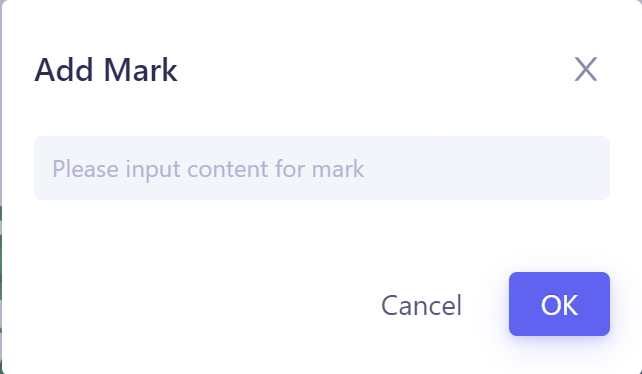
计算结果Mark标记:
-
支持对每一行数据进行标记,并支持搜索
-
点击Operation列中的show展示该任务的结果 → 打开的界面如右图所示。
-
-
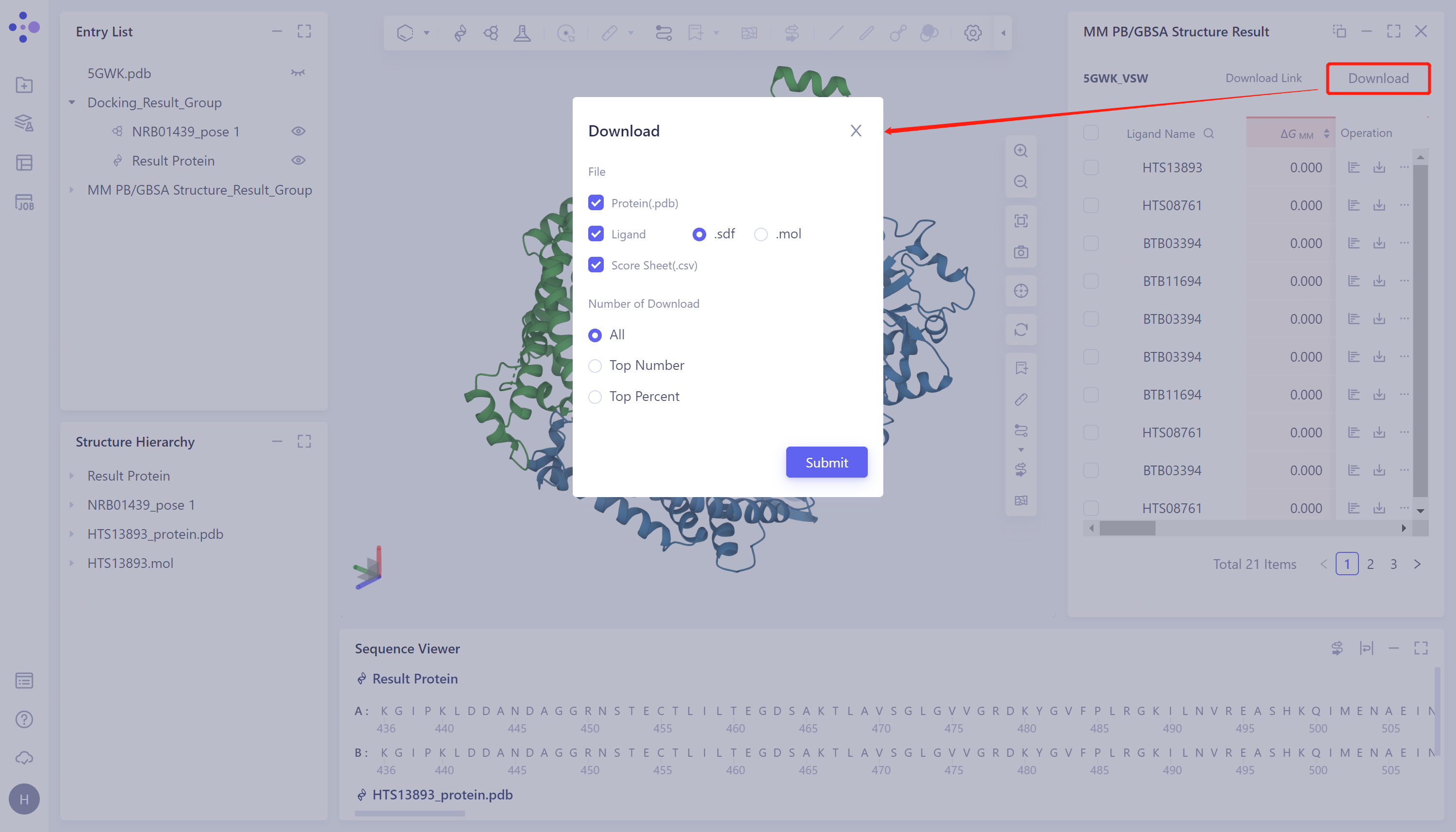
计算结果下载:
- 点击图中红色箭头处“Download”按钮,选择下载输出的参数
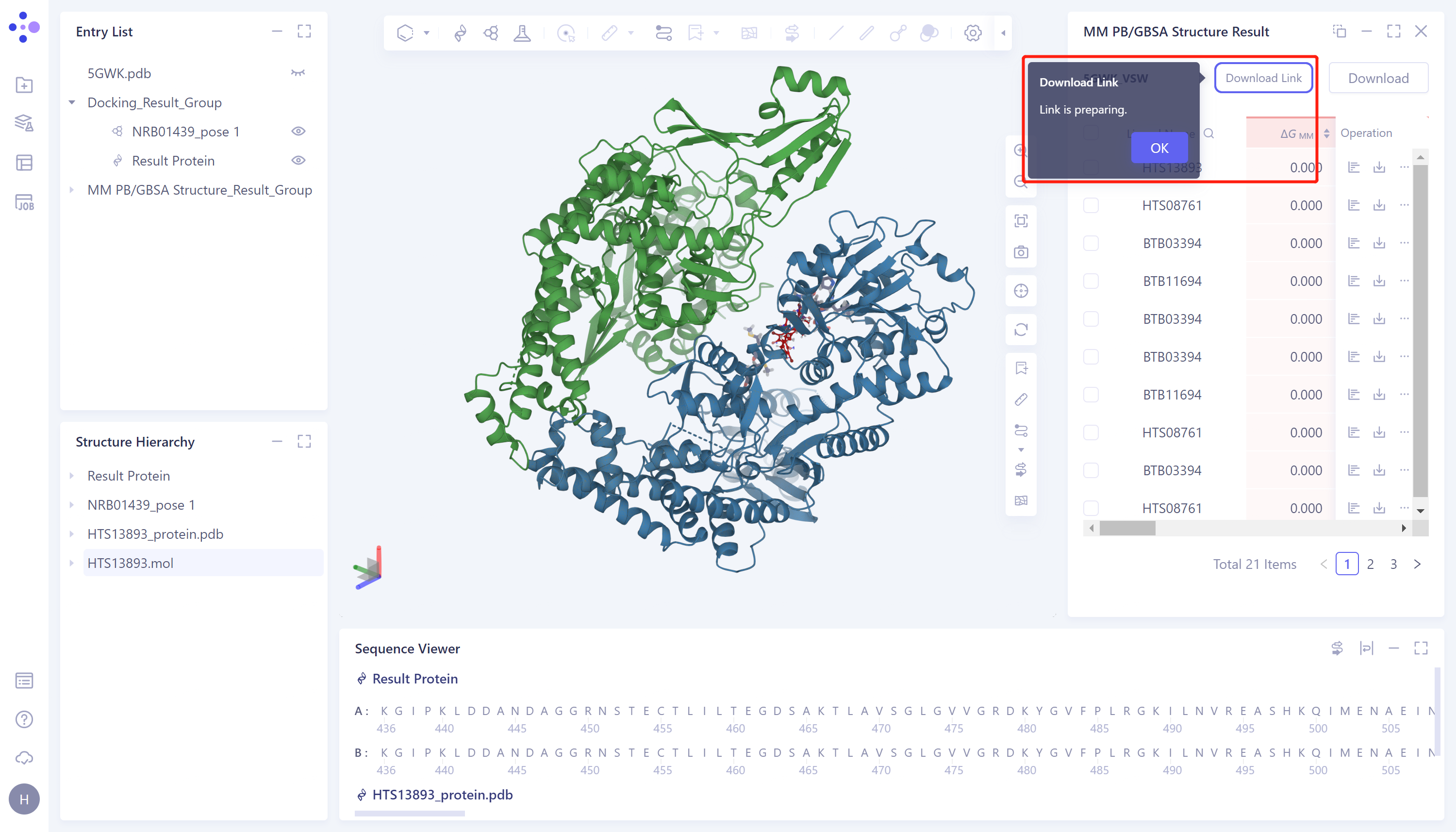
- 显示“Link is preparing”,等待加载完成
- 加载完成之后,点击“Download Link”按钮,点击URL链接下载计算结果。