Sequence BLAST
1. 前言
BLAST(Basic Local Alignment Search Tool)即“基于局部比对算法的搜索工具”,是生物信息学常用的工具软件,可将输入的核酸或蛋白质序列与数据库中的已知序列进行比对,获得序列相似度等信息,从而判断序列的来源或进化关系。BLAST广泛应用于生物信息学和分子生物学研究中,包括基因组注释、序列比对、基因家族分析、物种分类、蛋白质功能预测等方面。
Hermite平台的Sequence BLAST模块提供了蛋白质序列与数据库中已知序列比对的功能,支持从三种数据库中检索:Non-redundant protein sequences (nr)、UniProtKB/ Swiss-Prot (swissprot)、Protein Data Bank protein (pdb)。
2. 使用方法
2.1 入口
- 左侧通用菜单栏Menu → Function → Biomacromolecule �→ Sequence BLAST。
- 右侧出现Sequence BLAST的操作框(红框内所示),整体界面如下:
2.2 操作
-
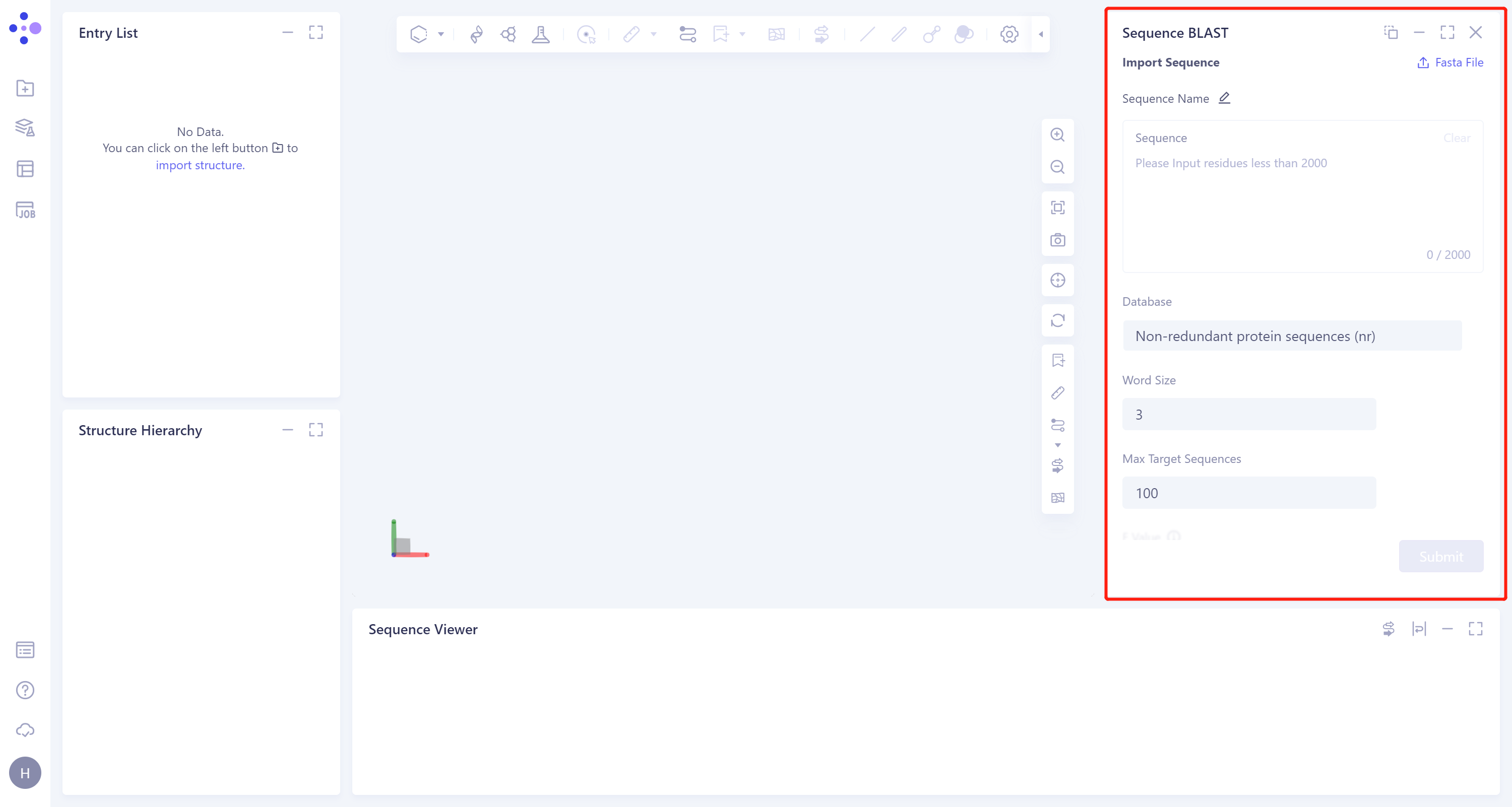
Import Srquence:上传序列,支持两种方式:
-
在Sequence框内输入序列;
-
支持 .fasta格式的文件上传。
注:输入的单条序列长度需低于2000。
-
-
Database:支持从三种数据库中检索:
-
Non-redundant protein sequences (nr);
-
UniProtKB/ Swiss-Prot (swissprot);
-
Protein Data Bank protein (pdb)。
-
-
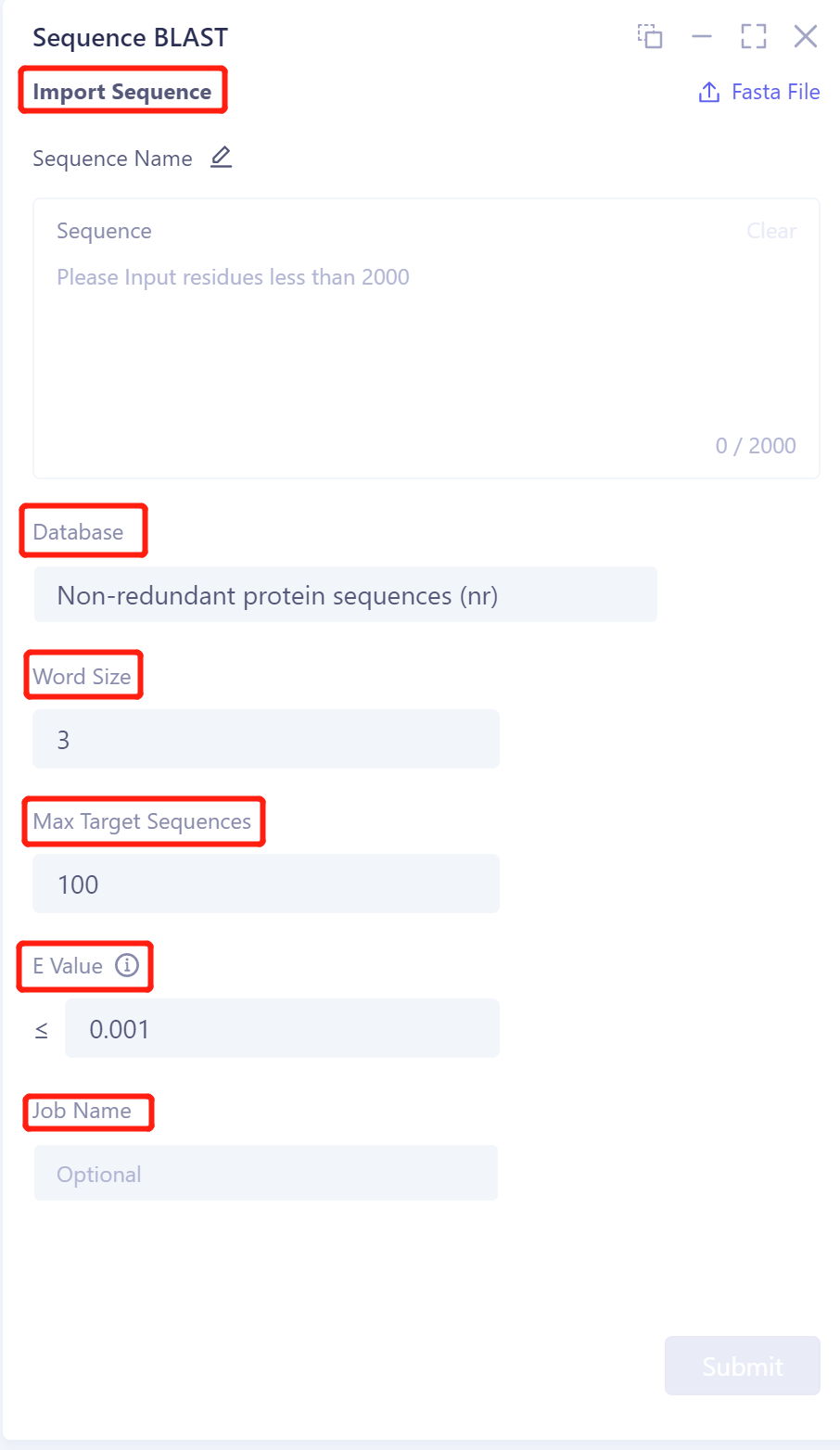
Word Size:比对长度,对于蛋白质搜索,可设置为2或3,参数默认为3;
- BLAST算法将查询序列分割成一系列具有字段长度的小的序列段进行数据库搜索,因此当此值越小得到的搜索结果越多,但假阳性也越多,服务器负担也越重。
-
Max Target Sequences:保留的最匹配的目标序列数;
- 支持输入50~1000。
-
E Value:表示仅仅因为随机性造成这一比对结果的可能次数,分值越低越好。
- 范围:0.001~1。
-
Job Name处对该任务进行命名。
-
Submit提交任务。
3. 结果分析
3.1 入口
-
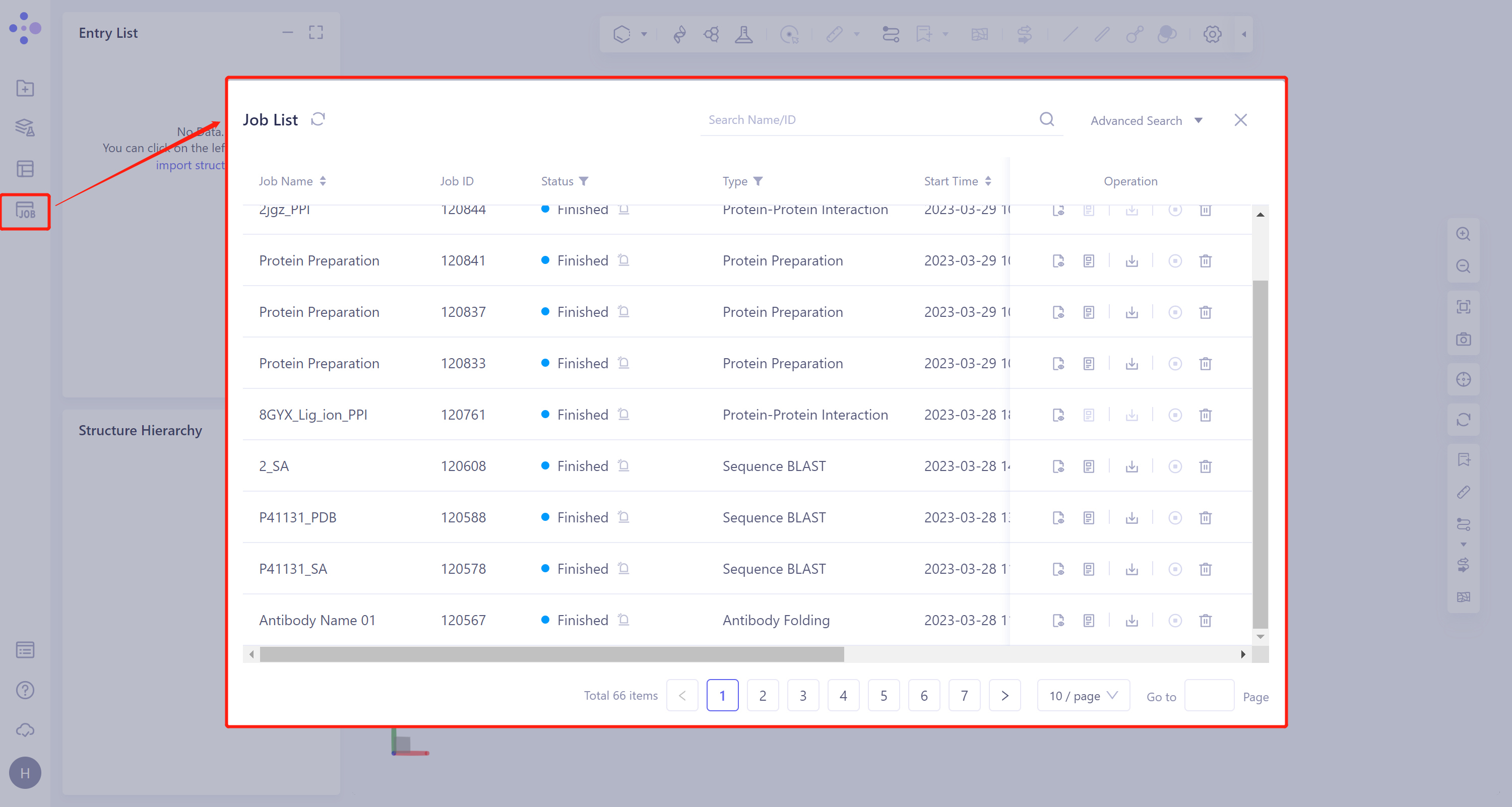
左侧通用菜单栏Menu Job → 找到所需任务。
- 可以通过搜索Job Name找到该任务,也可以通过Job Type的筛选找到。
3.2 操作
3.2.1 结果展示
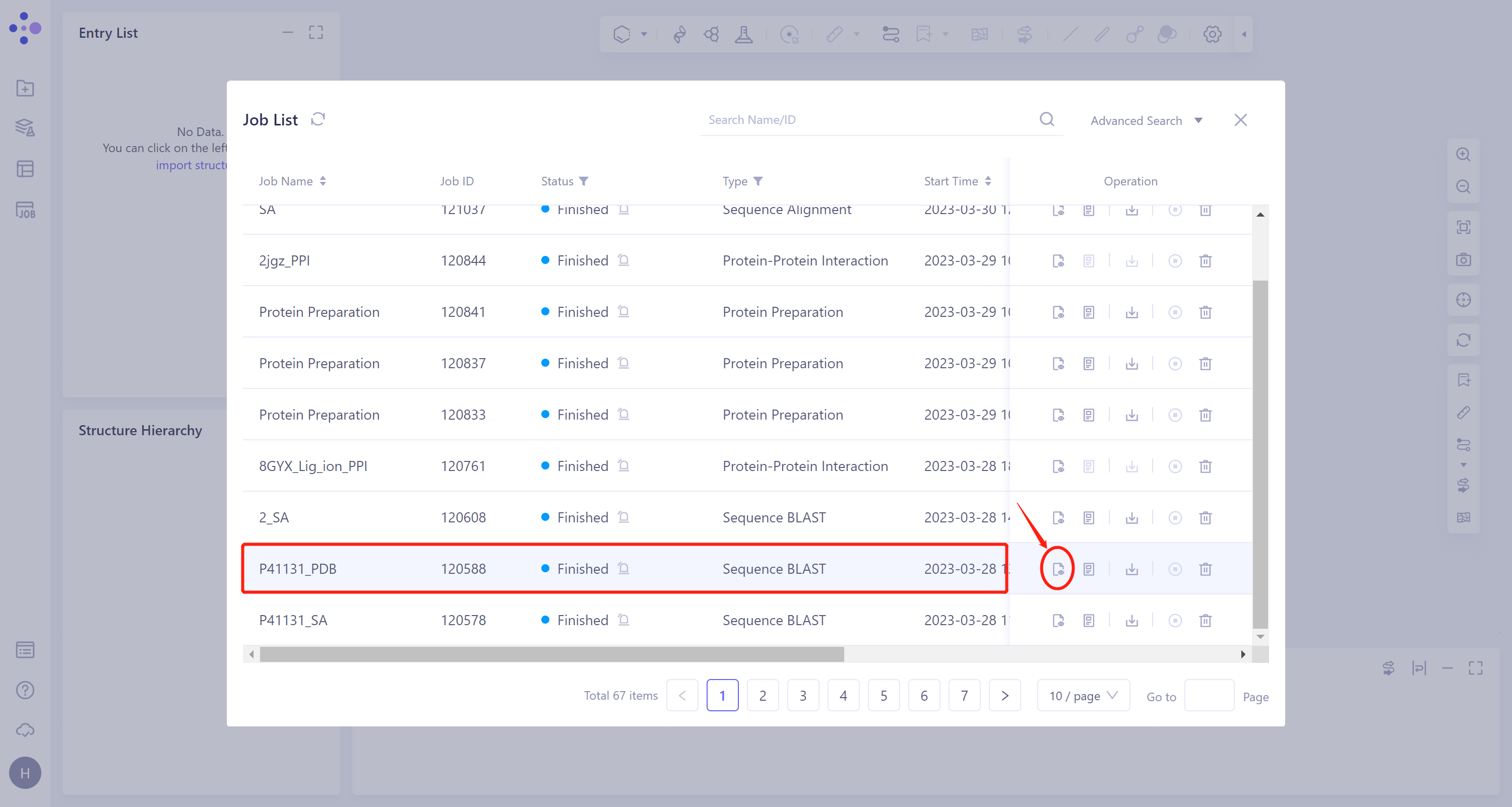
- 选择需要查看的任务,点击Operation列中的Show显示该任务的结果,界面如图所示。
 |  |
3.2.2 结果说明
-
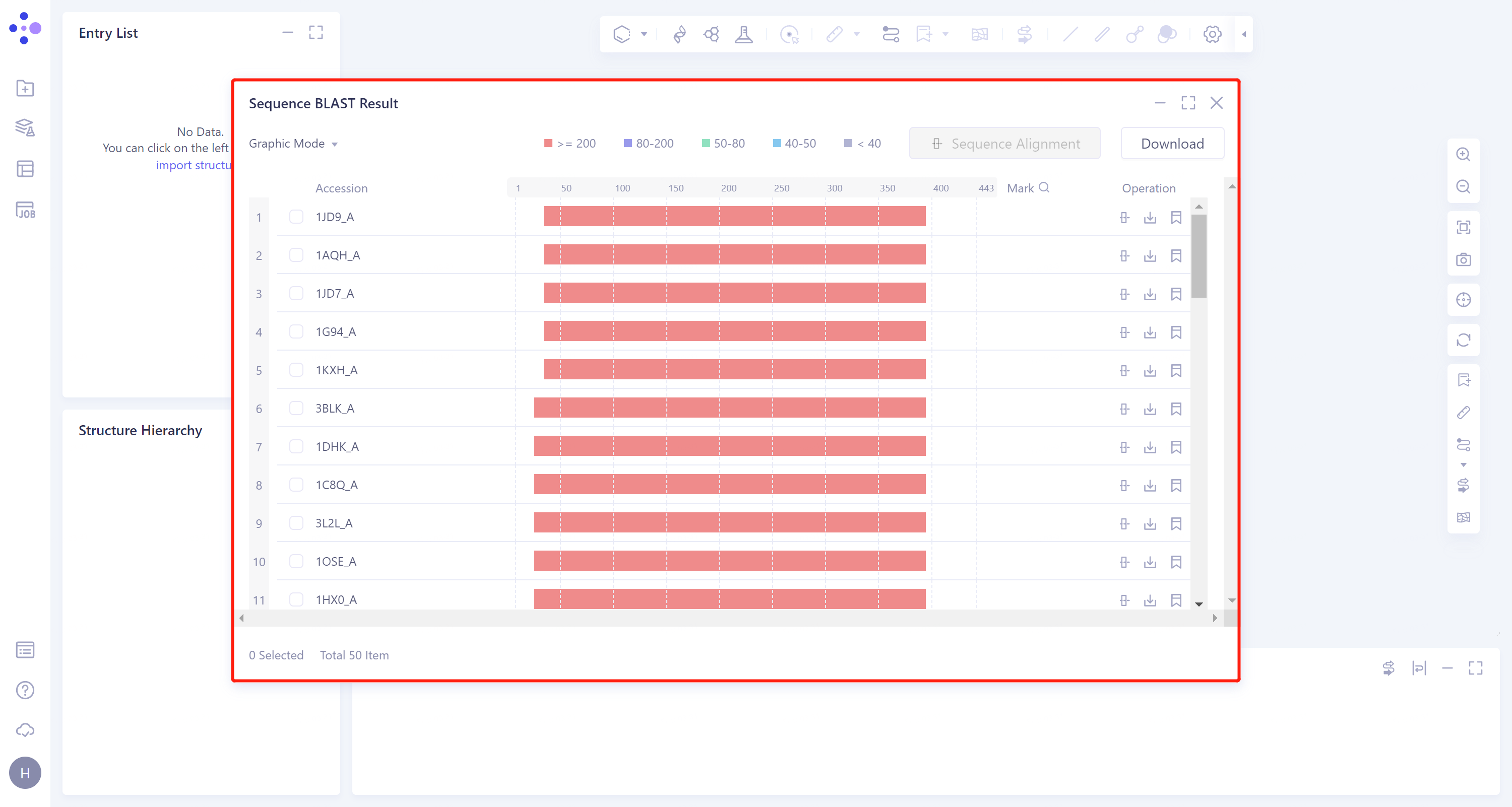
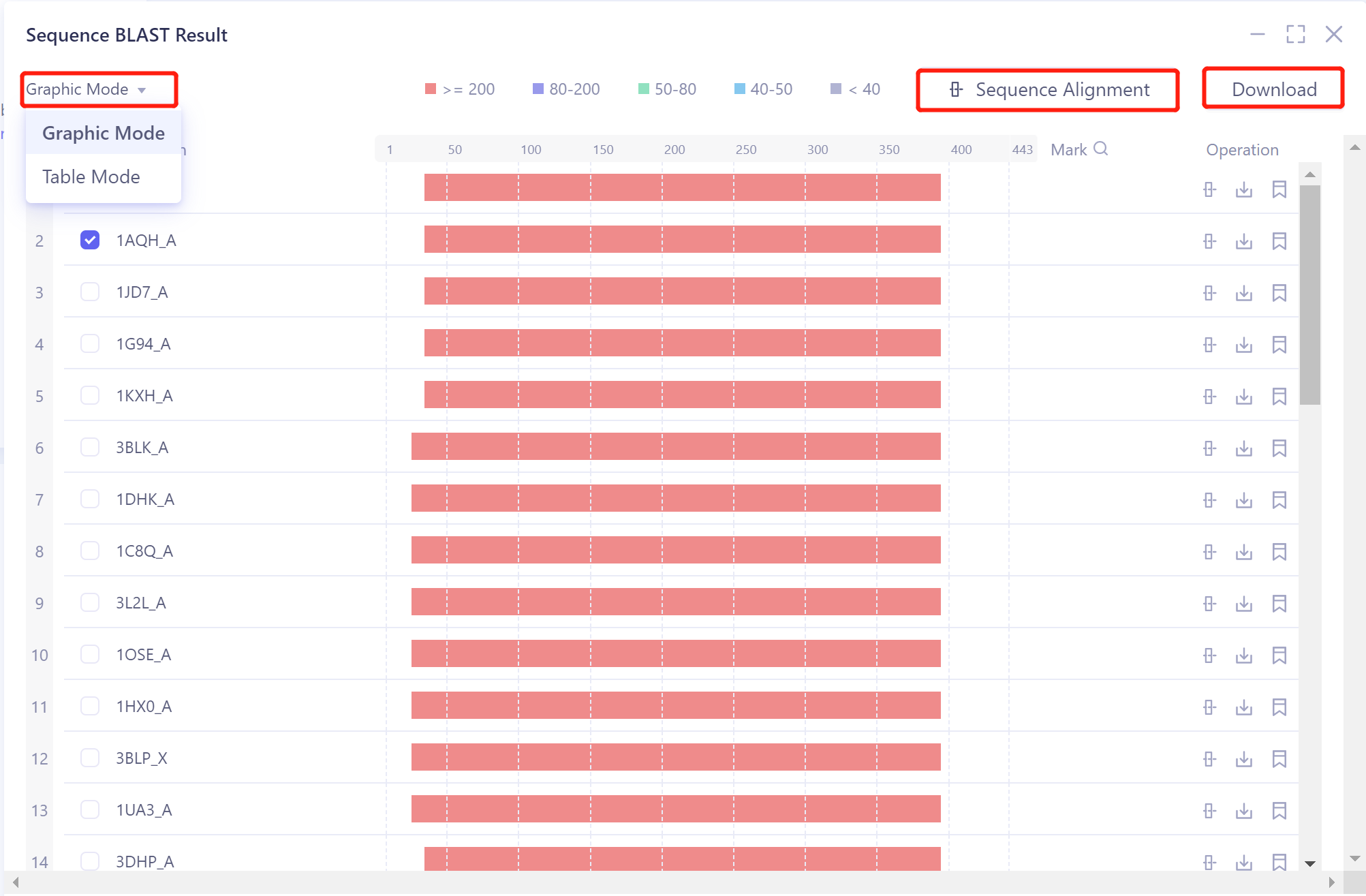
Graphic Mode:调节BLAST结果显示方式,有Graphic Mode和Table Mode两种显示方式,默认为Graphic Mode。
-
Sequence Alignment:对结果列表中选中的序列进行序列比对。
-
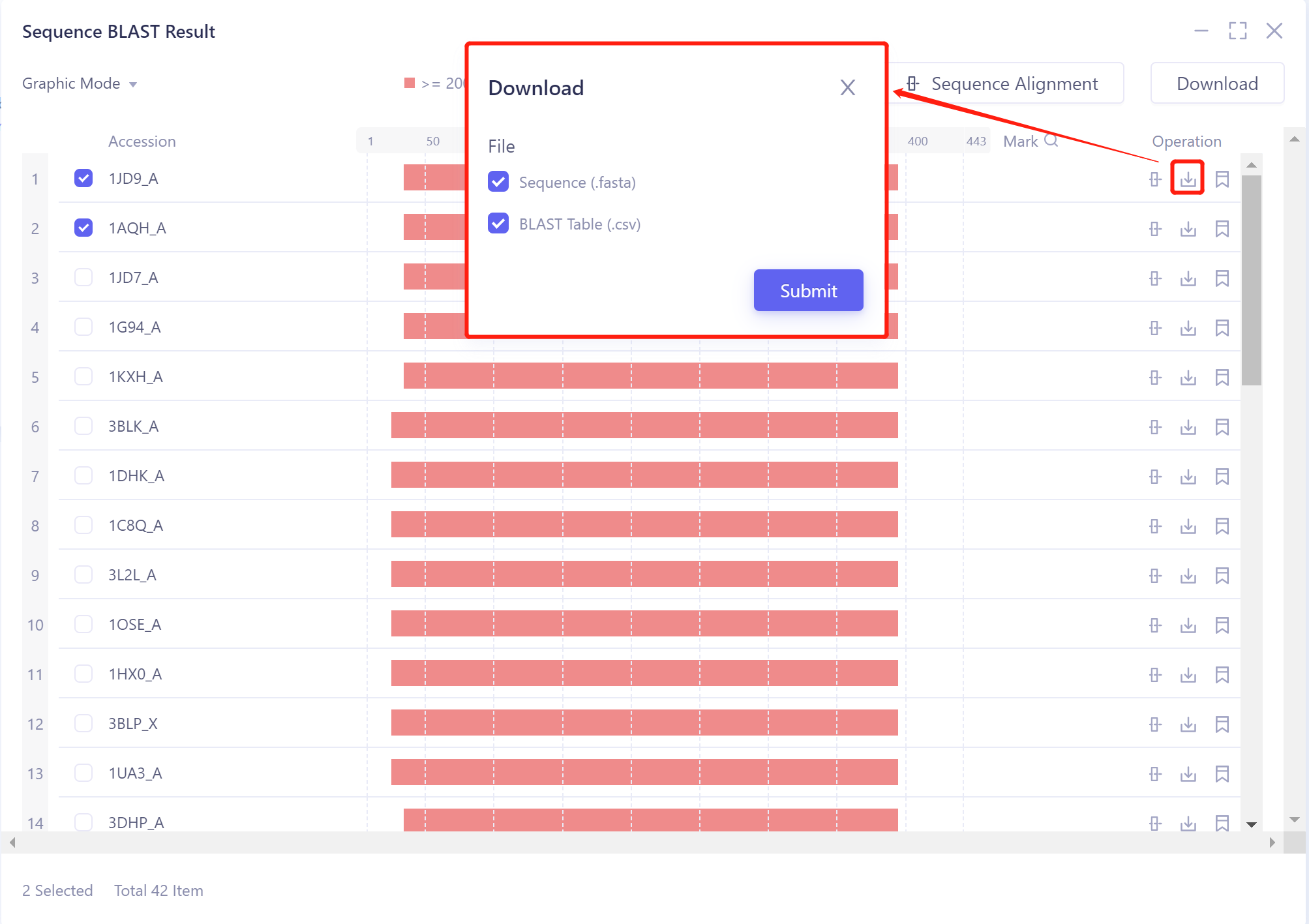
Download:下载结果列表中选中的序列的 .fasta 格式文件和BLAST结果的表格文件。
-
比对出的序列依据 bit scores 进行排序,并依据bit scores的值对比对出的序列使用不同颜色标注:红色(>= 200)、紫色 (80 ~ 200)、绿色 (50 ~ 80)、蓝色 (40 ~ 50)、灰色 (< 40)。
-
Operation下的操作:
1)Alignment:数据库检索到的某条序列与目标序列(输入序列)的序列比对,具体说明见Sequence Alignment V2023-3-15 编辑中。
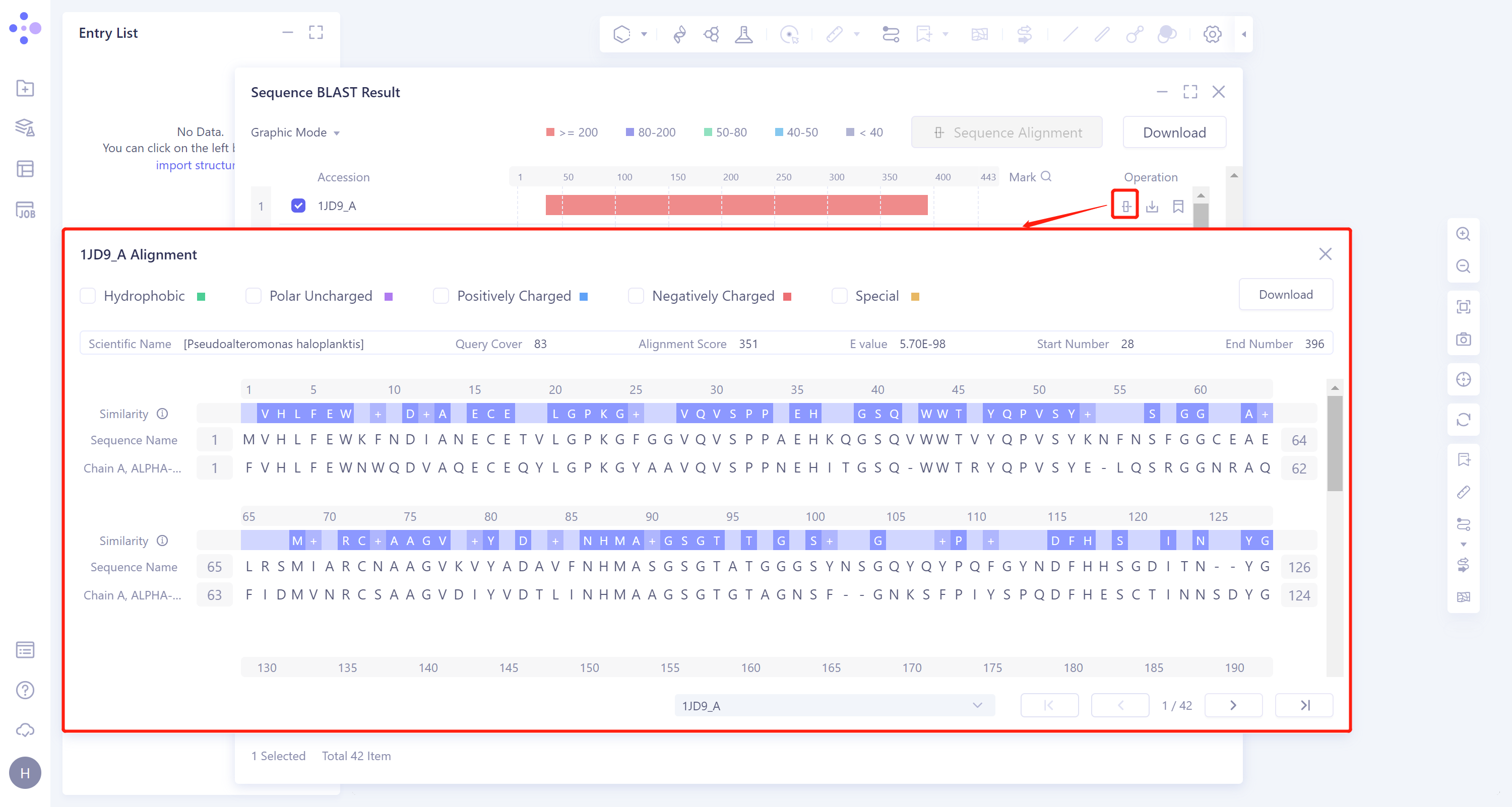 2)Download:下载某条比对序列的 .fasta 格式文件和BLAST的结果表格;
2)Download:下载某条比对序列的 .fasta 格式文件和BLAST的结果表格;
注:批量选择序列下载使用右上角Download,下载全部使用Job List界面中的Operation下的5个图标中的第3个(download)。
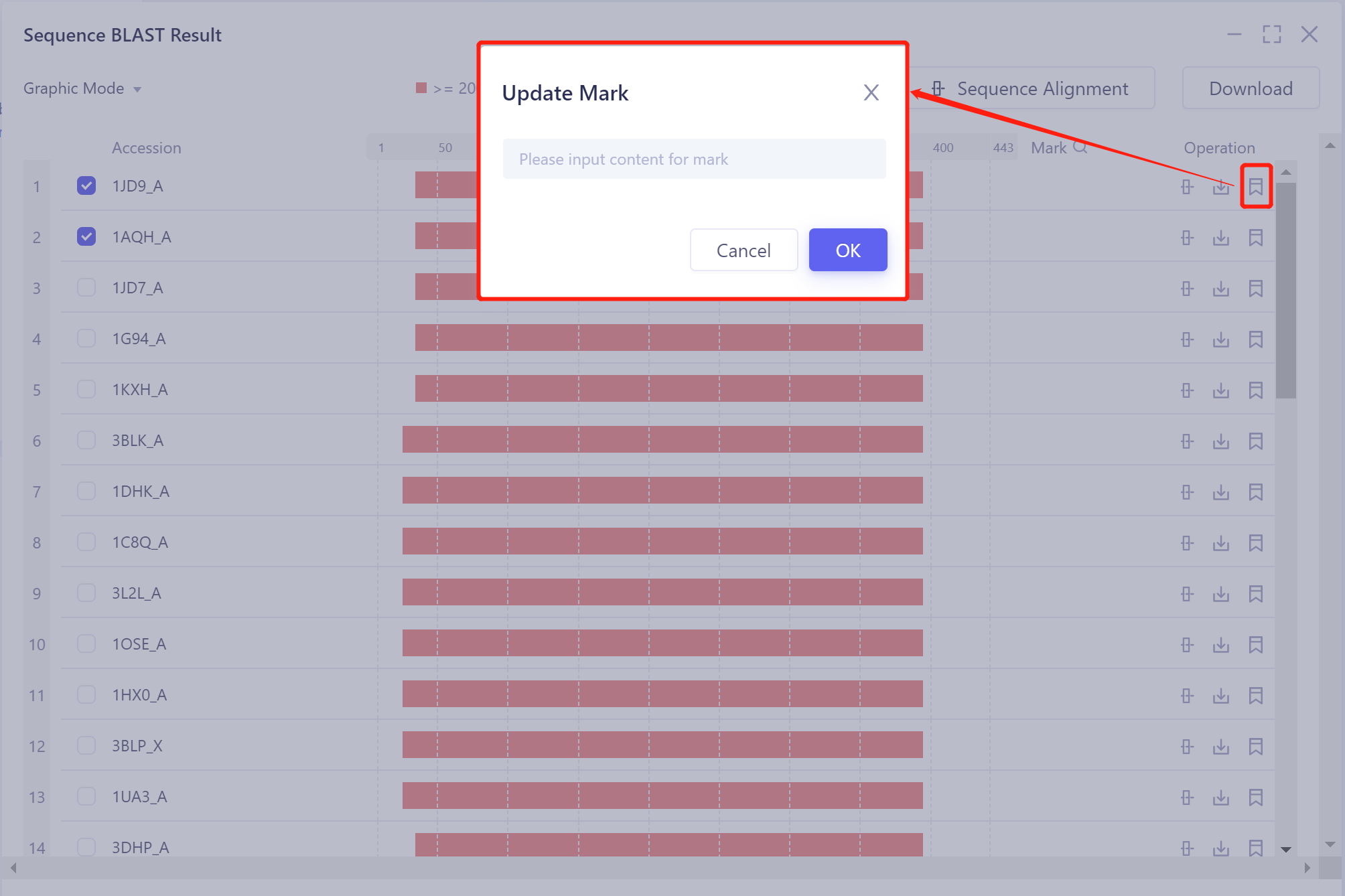
3)Mark:对某条序列进行标记;
注:标记内容会出现在左侧Mark一列中。